| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 100g |
|
||
| Other Sizes |
|

| 体外研究 (In Vitro) |
Probenecid 有效抑制 MRP1 和 MRP2 对 ATP 依赖性活性囊泡 N-乙基马来酰亚胺谷胱甘肽 (NEM-GS) 的吸收。随着有机阴离子浓度的增加,MRP1-ATP酶受到显着抑制。 MRP2的ATPase活性受丙磺舒(约KACT=250μM)、磺吡酮(KACT=300μM)和吲哚美辛(KACT=150μM)的影响,且ATPase激活甚至比NEM-GS更强。有机阴离子对 MRP2-ATP 酶的激活遵循钟形曲线,丙磺舒的最大值为 2 mM,磺吡酮的最大值为 800 μM,吲哚美辛的最大值为 400 μM [2]。 Probenecid 是 hTAS2R16、hTAS2R38 和 hTAS2R43 苦味受体的抑制剂。丙磺舒作用于 TAS2R 的一个子集,并通过新的变构作用机制进行抑制。丙磺舒也经常用于改善 GPCR 钙动员实验中的细胞信号传导。丙磺舒特异性抑制苦味受体 hTAS2R16 介导的细胞反应,并为使用非竞争性(变构)机制与该 GPCR 直接接触提供分子和药理学证据 [3]。
|
||
|---|---|---|---|
| 体内研究 (In Vivo) |
与饲喂盐水的对照小鼠相比,丙磺舒增加了 WT 小鼠的收缩力,如射血分数 (EF) 所示。在 75 mg/kg 及更高剂量的所有剂量下,在推注后 5 分钟内观察到收缩力增加(75 mg/kg、100 mg/kg 和 200 mg/kg 时的峰值变化分别为 5.26±3.35、8.40±2.80 和分别为7.32±2.52)。估计 EC50 为 49.33 mg/kg,在总共 30 分钟内以 5 分钟为间隔评估的收缩性变化显示出收缩性的剂量依赖性增加。在较长时间内接受检查的患者中,EF 保持升高至少一个小时(n=5,200 mg/kg IV)(EF 相对于基线的平均升高为 8.9±2.57)[1]。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Excreted principally in the urine as monoacyl glucuronide and unchanged drug. Alkalinization of urine increases renal probenecid excretion. PROBENECID IS COMPLETELY ABSORBED AFTER ORAL ADMIN. PEAK PLASMA CONCN ARE REACHED IN 2-4 HR. THE HALF-LIFE OF THE DRUG IN PLASMA IS DOSE DEPENDENT AND VARIES FROM LESS THAN 5 HR TO MORE THAN 8 HR. BETWEEN 85 & 95% OF DRUG IS BOUND TO PLASMA ALBUMIN, LARGELY TO ALBUMIN. SMALL UNBOUND PORTION GAINS ACCESS TO GLOMERULAR FILTRATE; A MUCH LARGER PORTION IS ACTIVELY SECRETED BY PROXIMAL TUBULE. IN SPITE OF ITS LOW PKA (3.4), HIGH LIPID SOLUBILITY OF UNDISSOCIATED FORM RESULTS IN VIRTUALLY COMPLETE ABSORPTION BY BACK DIFFUSION UNLESS URINE IS MARKEDLY ALKALINE. SMALL AMOUNT OF PROBENECID GLUCURONIDE APPEARS IN URINE. ... /ORG ACID CMPD SUCH AS PROBENECID /ARE/ NOT TAKEN UP SO AVIDLY BY /PARENCHYMATOUS OR RETICULO-ENDOTHELIAL TISSUES/ & EXHIBIT HIGHER PLASMA CONCN ... . For more Absorption, Distribution and Excretion (Complete) data for PROBENECID (8 total), please visit the HSDB record page. Metabolism / Metabolites YIELDS P-DIPROPYLSULFAMOYLBENZOYL-BETA-D-GLUCURONIC ACID; P-(2-HYDROXYPROPYL N-PROPYLSULFAMOYL) BENZOIC ACID; P-(3-HYDROXYPROPYL N-PROPYLSULFAMOYL) BENZOIC ACID; & P-PROPYLSULFAMOYLBENZOIC ACID IN MAN. /FROM TABLE/ STRUCTURES OF ALL OF METAB OF PROBENECID IN RAT BILE & HUMAN URINE HAVE BEEN ELUCIDATED. PROPIONIC ACID HAS NOW BEEN IDENTIFIED AS ANOTHER PROBENECID METAB. MAJOR METABOLIC PATHWAYS INVOLVE SIDE-CHAIN OXIDATION & GLUCURONIDE CONJUGATION ... . ... BETA-GLUCURONIDES OF 2- & 3-HYDROXYLATED METAB & ACYL GLUCURONIDE OF PROBENECID PER SE HAVE NOW BEEN IDENTIFIED. ... THERE IS CONSIDERABLE SPECIES DIFFERENCE IN METABOLISM. IN RATS & MONKEYS OXIDATION IS FAVORED. ... IN DOGS CONJUGATION ... /IS/ MAJOR PATHWAY, WHEREAS IN MAN, OXIDATIVE ... PATHWAY ... IS AS IMPORTANT AS GLUCURONIDATION. CHRONIC ADMIN OF DRUGS NOT ONLY STIMULATES METAB OF OTHER CMPD, BUT IN SOME INSTANCES PHARMACOLOGICAL OR TOXIC EFFECT OF A DRUG WHEN GIVEN CHRONICALLY, DIMINISHES, BECAUSE IT STIMULATES ITS OWN METABOLISM EXAMPLE OF DRUG THAT EXERT THIS EFFECT IN DOGS ... /IS/ ... PROBENECID ... . For more Metabolism/Metabolites (Complete) data for PROBENECID (6 total), please visit the HSDB record page. Biological Half-Life 6-12 hours THE HALF-LIFE OF /PROBENECID/ IN PLASMA IS DOSE DEPENDENT AND VARIES FROM LESS THAN 5 HR TO MORE THAN 8 HR ... . Following oral administration of 2 g of probenecid, plasma half-life of the drug ranges from 4-17 hr; the half-life decreases as the dose decreases from 2 g to 500 mg. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
There are no reports on the frequency of liver test abnormalities during probenecid therapy, but they are probably rare as the drug is largely secreted unchanged in the urine. A single case report of a severe hypersensitivity reaction from probenecid and rechallenge with a rapid and severe recurrence of jaundice was reported over 50 years ago. As is typical for hypersensitivity reactions, the onset was within days of starting probenecid and was accompanied by fever and rash. Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Limited information indicates that maternal doses of probenecid up to 2 grams daily produce low levels in milk and would not be expected to cause any adverse effects in breastfed infants, especially if the infant is older than 2 months. In animal studies, probenecid increased the breastmilk excretion of cimetidine, possible via an interaction with an active transport mechanism in the breast. The implications of enhanced excretion of drugs given with probenecid for nursing mothers and their infants has not been studied; however, only a few drugs are known to undergo active transport into breastmilk. ◉ Effects in Breastfed Infants A woman with mastitis received 3 days of intravenous cephalothin, followed by 16 days of probenecid 500 mg and cephalexin 500 mg 4 times daily for 16 days. Her infant developed green liquid stools, severe diarrhea, discomfort and crying. The authors judged the effects to be probably related to the cephalothin and cephalexin in milk rather than the probenecid. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding 75-95% |
||
| 参考文献 |
Cell Calcium.1989 Apr;10(3):171-80;Cardiovasc Toxicol.2012 Mar;12(1):1-9.
|
||
| 其他信息 |
Probenecid appears as odorless white or almost white crystalline powder. Slightly bitter taste; pleasant aftertaste. (NTP, 1992)
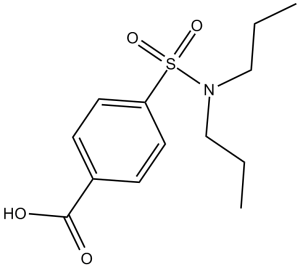
Probenecid is a sulfonamide in which the nitrogen of 4-sulfamoylbenzoic acid is substituted with two propyl groups. It has a role as a uricosuric drug. It is a sulfonamide and a member of benzoic acids. The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy. Probenecid is a uricosuric agent used for the treatment of gout usually in combination with other agents. Probenecid has been associated with minor serum aminotransferase elevations and very rarely with hypersensitivity reactions which, even more rarely, can be accompanied by acute liver injury. Probenecid is a benzoic acid derivative with antihyperuricemic property. Probenecid competitively inhibits the active reabsorption of urate at the proximal tubule in the kidney thereby increasing urinary excretion of uric acid and lowering serum urate concentrations. This prevents urate deposition and promotes resolution of existing urate deposits. In addition, probenecid modulates the transport of organic acids and acidic drugs at the proximal and distal renal tubule, thereby increasing the drug serum concentration. The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy. See also: Colchicine; probenecid (component of); Ampicillin/ampicillin trihydrate; probenecid (component of). Drug Indication For the reduction of serum uric acid concentrations in chronic gouty arthritis and tophaceous gout in patients with frequent disabling gout attacks. Has also been effectively used to promote uric acid excretion in hyperuricemia secondary to the administration of thiazide and related diuretics. Mechanism of Action Probenecid inhibits the tubular reabsorption of urate, thus increasing the urinary excretion of uric acid and decreasing serum urate levels. Probenecid may also reduce plasma binding of urate and inhibit renal secretion of uric acid at subtherapeutic concentrations. The mechanism by which probenecid inhibits renal tubular transport is not known, but the drug may inhibit transport enzymes that require a source of high energy phosphate bonds and/or nonspecifically interfere with substrate access to protein receptor sites on the kidney tubules. IN HIGHER DOSES THAN ARE REQUIRED FOR URICOSURIC EFFECT, PROBENECID ALSO INHIBITS TRANSPORT OF ORG ACIDS AT OTHER SITES, IE, TRANSPORT SYSTEM THAT REMOVES ORG ACIDS FROM CEREBROSPINAL FLUID. IT INHIBITS TUBULAR REABSORPTION OF URATE, THUS INCR URINARY EXCRETION OF URIC ACID & DECR SERUM URIC ACID LEVELS. Probenecid is a renal tubular blocking agent. The drug competitively inhibits active reabsorption of uric acid at the proximal convoluted tubule, thus promoting urinary excretion of uric acid and reducing serum urate concentrations. Probenecid may reduce plasma protein binding of urate and, in subtherapeutic doses, may inhibit renal secretion of uric acid. In healthy individuals, probenecid has no effect on the glomerular filtration rate or on the tubular reabsorption of normal urinary constituents such as glucose, arginine, urea, sodium, potassium, chloride, or phosphate. At the proximal and distal tubules, probenecid competitively inhibits the secretion of many weak organic acids including penicillins, most cephalosporins, and some other beta-lactam antibiotics. In general, the net effect of probenecid on the plasma concentration of weak acids depends on the ratio of the amount of organic acid secreted by the kidneys to that amount filtered at the glomeruli. Thus, probenecid substantially increases plasma concentrations of acidic drugs eliminated principally by renal secretion, but increases plasma concentrations only slightly if the drug is eliminated mainly by filtration. Plasma concentrations of penicillins are often more than doubled by probenecid; the concentration of penicillin in the CSF is also increased. Probenecid also substantially increases plasma concentrations of most cephalosporins and some other beta-lactam antibiotics. In addition, half-lives of the penicillins and cephalosporins are prolonged and their volumes of distribution may be reduced by probenecid. ... The cellular mechanism(s) responsible for the inhibition of renal tubular transport by probenecid is not known. The drug may inhibit transport enzymes that require a source of high energy phosphate bonds and/or nonspecifically interfere with substrate access to protein receptor sites on the kidney tubules. CSF concentrations of 5-hydroxyindoleacetic acid, homovanillic acid, cyclic adenosine monophosphate, and 4-hydroxy-3-methoxyphenylglycol are elevated following administration of probenecid. It has been proposed that probenecid blocks the active transport of these organic acids from the CSF into blood. Probenecid-induced elevations of homovanillic acid (a dopamine metabolite) in the CSF of patients with parkinsonian syndrome and of 5-hydroxyindoleacetic acid(a metabolite of serotonin) in the CSF of mentally depressed patients are substantially lower than those in healthy patients. |
| 分子式 |
C13H19NO4S
|
|
|---|---|---|
| 分子量 |
285.36
|
|
| 精确质量 |
285.103
|
|
| CAS号 |
57-66-9
|
|
| 相关CAS号 |
Probenecid;57-66-9
|
|
| PubChem CID |
4911
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.2±0.1 g/cm3
|
|
| 沸点 |
438.0±47.0 °C at 760 mmHg
|
|
| 熔点 |
194-196°C
|
|
| 闪点 |
218.7±29.3 °C
|
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
|
| 折射率 |
1.542
|
|
| LogP |
3.3
|
|
| tPSA |
83.06
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
5
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
19
|
|
| 分子复杂度/Complexity |
374
|
|
| 定义原子立体中心数目 |
0
|
|
| InChi Key |
DBABZHXKTCFAPX-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C13H19NO4S/c1-3-9-14(10-4-2)19(17,18)12-7-5-11(6-8-12)13(15)16/h5-8H,3-4,9-10H2,1-2H3,(H,15,16)
|
|
| 化学名 |
4-(dipropylsulfamoyl)benzoic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.5 mg/mL (8.76 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (8.76 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5043 mL | 17.5217 mL | 35.0435 mL | |
| 5 mM | 0.7009 mL | 3.5043 mL | 7.0087 mL | |
| 10 mM | 0.3504 mL | 1.7522 mL | 3.5043 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 ER degrader 10
ER degrader 10
 VU0546110
VU0546110
 hERG-IN-2
hERG-IN-2
 KNa1.1-IN-2
KNa1.1-IN-2
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
