| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
JAK/Janus kinases; Ilunocitinib targets JAK1, JAK2, and TYK2 (tyrosine kinase 2); no specific IC50, Ki, or EC50 values are provided in the text.
|
|---|---|
| 体外研究 (In Vitro) |
Ilunocitinib 在体外对 JAK1、JAK2 和 TYK2 表现出高效抑制效力,未发表数据显示其在酶活性实验中能强力抑制这些激酶,表明其可有效阻断炎症和瘙痒相关的细胞因子信号通路。 [1]
蛋白激酶(PKs)是一组调节多种重要生物过程的酶,包括细胞生长、存活和分化、器官形成和形态发生、新生血管形成、组织修复和再生等。蛋白激酶通过催化蛋白质(或底物)的磷酸化,从而调节底物在各种生物环境下的细胞活性,发挥其生理功能。除了在正常组织/器官中的功能外,许多蛋白激酶在包括癌症在内的许多人类疾病中也起着更特殊的作用。蛋白激酶的一个子集(也被称为致癌蛋白激酶),当失调时,可引起肿瘤的形成和生长,并进一步促进肿瘤的维持和进展。到目前为止,致癌蛋白激酶是癌症干预和药物开发中最大和最具吸引力的蛋白质靶点之一。Janus激酶(JAK)家族在参与免疫应答的细胞因子依赖的增殖和功能调节中发挥作用。目前,已知的哺乳动物JAK家族成员有四种:JAKl(也称为Janus激酶- 1)、JAK2(也称为Janus激酶-2)、JAK3(也称为Janus激酶、白细胞;JAKL;L-JAK和Janus激酶-3)和TYK2(也称为蛋白酪氨酸激酶T)。JAK蛋白的大小从120到140 kDa不等,包含7个保守的JAK同源(JH)结构域;其中一个是功能性催化激酶结构域,另一个是假激酶结构域,可能具有调节功能和/或作为STATs的对接位点。 在JAK激酶水平上阻断信号转导有望开发炎症性疾病、自身免疫性疾病、骨髓增殖性疾病和人类癌症的治疗方法,仅举几例。JAK激酶的抑制也被设想对患有皮肤免疫疾病(如牛皮癣)和皮肤致敏的患者具有治疗益处。因此,Janus激酶或相关激酶的抑制剂被广泛寻找,一些出版物报道了有效的化合物类别。例如,某些JAK抑制剂,包括吡咯吡啶和吡咯嘧啶,在美国报道。第11/637,545号,2006年12月12日提交。 |
| 体内研究 (In Vivo) |
在一项纳入 338 只犬特应性皮炎(cAD)患犬的随机盲法试验中,Ilunocitinib(0.6–0.8 mg/kg,每日一次)能快速减轻瘙痒和皮肤病变。第 0 天至第 14 天,瘙痒视觉模拟评分(PVAS)均值下降与奥拉替尼相似,两组较基线均降低约 50%。第 28 天至第 112 天,Ilunocitinib 组的平均 PVAS 评分显著低于奥拉替尼组(p < 0.003),且在整个治疗期间持续改善。此外,更多 Ilunocitinib 治疗犬达到瘙痒临床缓解(定义为 PVAS < 2),第 112 天时达 77%,而奥拉替尼组为 53%(第 56 天和第 112 天 p < 0.04)。
对于皮肤病变(使用犬特应性皮炎范围和严重程度指数 CADESI-04 评估),Ilunocitinib 显示快速改善:第 14 天评分降至基线一半以下。第 28 天至第 112 天,Ilunocitinib 组的平均 CADESI-04 评分显著低于奥拉替尼组(p < 0.023)。第 112 天时,69% 的 Ilunocitinib 治疗犬达到皮肤病变临床缓解(CADESI-04 < 10),数值上高于奥拉替尼组(64%)。其抗炎作用归因于阻断瘙痒-抓挠循环及抑制 JAK-STAT 通路促炎细胞因子信号。 主人和研究者对总体治疗响应(ORTT 和 IRTT)的评估从第 28 天起显著更优(p ≤ 0.002),表明临床改善和生活质量提升更佳。[1] Ilunocitinib (LY-3411067)是一种新型有效的Janus激酶抑制剂,作为兽药已被批准。 |
| 动物实验 |
Dogs were randomized to receive Ilunocitinib tablets orally at 0.6–0.8 mg/kg body weight once daily for up to 112 days. Tablets were administered with or without food, at the owner's discretion, and given at approximately the same time each day. The study was a prospective, double-blinded, randomized, positive-controlled field trial conducted at 25 veterinary clinics across Germany, Hungary, Ireland, and Portugal. Efficacy assessments included owner-reported PVAS and investigator-assessed CADESI-04 at baseline (Day 0), Day 14 (±2 days), Day 28 (±2 days), Day 56 (±3 days), and optionally at Day 84 (±3 days) and Day 112 (±3 days) for dogs in the continuation phase. Safety monitoring included physical examinations, hematology, and serum chemistry at all visits. [1]
|
| 毒性/毒理 (Toxicokinetics/TK) |
Ilunocitinib exhibited a favorable safety profile over 112 days, with adverse events (AEs) similar to oclacitinib. Digestive tract disorders were most common, including emesis (13.6% incidence) and diarrhea. Systemic disorders occurred in 5.3% of dogs, and skin/appendage disorders in 5.9%. Most AEs were mild and assessed as possibly treatment-related. Hematological changes included decreases in leucocyte counts, primarily neutrophils and eosinophils, but values remained within normal laboratory reference ranges. No clinically relevant abnormalities were observed in serum biochemistry. Serious adverse events (e.g., seminoma) were not treatment-related. [1]
|
| 参考文献 | |
| 其他信息 |
Ilunocitinib is a novel Janus kinase inhibitor (JAKi) designed for once-daily oral administration to control pruritus and skin lesions in dogs with atopic dermatitis. Its mechanism involves potent inhibition of JAK1, JAK2, and TYK2, reducing signaling of pro-inflammatory cytokines (e.g., IL-31) via the JAK-STAT pathway, which is central to cAD pathogenesis. Clinical relevance includes superior long-term efficacy over oclacitinib, with higher rates of clinical remission and reduced caregiver burden due to consistent dosing. No FDA alerts or specific drug interactions were reported, and it is indicated for managing cAD manifestations. [1]
Background: Janus kinase inhibitors (JAKi) have been shown to reduce pruritus and improve associated inflammatory skin lesions in canine atopic dermatitis (cAD). Objective: To evaluate the efficacy and safety of ilunocitinib, in comparison to oclacitinib, for the control of cAD in a randomised, blinded trial. Animals: Three-hundred-and-thirty-eight dogs with cAD. Materials and methods: Dogs were randomised to receive oclacitinib (0.4-0.6 mg/kg twice daily for 14 days; then once daily) or ilunocitinib (0.6-0.8 mg/kg once daily), for up to 112 days. Owners assessed pruritus using an enhanced Visual Analog Scale (PVAS). Investigators assessed skin lesions using the Canine Atopic Dermatitis Extent and Severity Index, 4th interaction (CADESI-04). Results: Reduction in pruritus and CADESI-04 scores was similar for both treatment groups from Day (D)0-D14. PVAS scores increased between D14 and D28 for oclacitinib and decreased for ilunocitinib. On D28 to D112, mean PVAS and CADESI-04 scores were significantly lower for ilunocitinib compared to oclacitinib (p ≤ 0.003 and p ≤ 0.023, respectively). On D28 to D112, a greater number of ilunocitinib-treated dogs achieved clinical remission of pruritus (i.e. PVAS score <2). Subjective assessment of overall response was significantly better for ilunocitinib on D28 to D112 (p ≤ 0.002). Both drugs demonstrated similar safety throughout the study. Conclusions and clinical relevance: Ilunocitinib rapidly and safely controlled signs of cAD. Ilunocitinib demonstrated significantly better control of pruritus and skin lesions compared to oclacitinib, with more dogs achieving clinical remission of pruritus. [1] |
| 分子式 |
C17H17N7O2S
|
|---|---|
| 分子量 |
383.4276
|
| 精确质量 |
383.116
|
| 元素分析 |
C, 53.25; H, 4.47; N, 25.57; O, 8.35; S, 8.36
|
| CAS号 |
1187594-14-4
|
| PubChem CID |
44231134
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
-0.3
|
| tPSA |
129
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
734
|
| 定义原子立体中心数目 |
0
|
| SMILES |
S(C1([H])C([H])([H])C1([H])[H])(N1C([H])([H])C(C([H])([H])C#N)(C1([H])[H])N1C([H])=C(C2=C3C([H])=C([H])N([H])C3=NC([H])=N2)C([H])=N1)(=O)=O
|
| InChi Key |
RVOUEXFKIYNODQ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C17H17N7O2S/c18-5-4-17(9-23(10-17)27(25,26)13-1-2-13)24-8-12(7-22-24)15-14-3-6-19-16(14)21-11-20-15/h3,6-8,11,13H,1-2,4,9-10H2,(H,19,20,21)
|
| 化学名 |
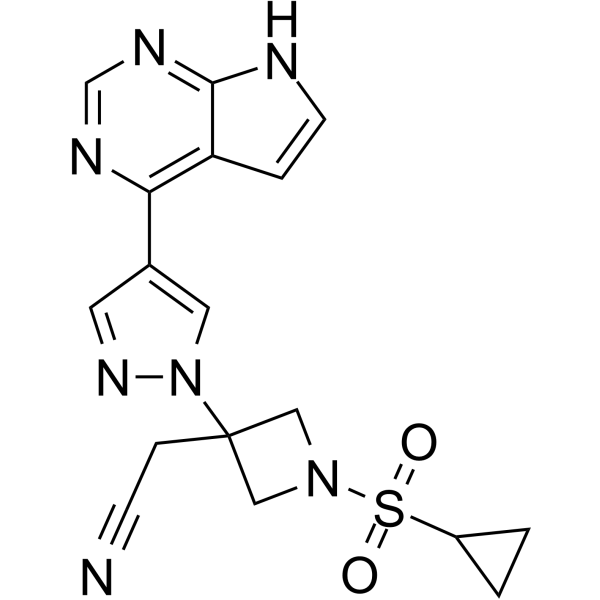
2-[1-cyclopropylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile
|
| 别名 |
Ilunocitinib; 1187594-14-4; Ilunocitinib [USAN]; LY3411,067; N3TB5AH8B9; LY3411,067; LY3411067; 2-[1-cyclopropylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile; ILUNOCITINIB [INN]; N3TB5AH8B9; UNII-N3TB5AH8B9; LY-3411067; WHO 11894;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~250 mg/mL (~652.01 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (5.42 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (5.42 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (5.42 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6080 mL | 13.0402 mL | 26.0804 mL | |
| 5 mM | 0.5216 mL | 2.6080 mL | 5.2161 mL | |
| 10 mM | 0.2608 mL | 1.3040 mL | 2.6080 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 463611831
463611831
