| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
AT-56 is an orally active and selective inhibitor of lipocalin-type prostaglandin D synthase (L-PGDS) [1]. It inhibits human and mouse L-PGDSs in a concentration-dependent manner but does not affect the activities of hematopoietic PGD synthase (H-PGDS), cyclooxygenase-1 (COX-1), cyclooxygenase-2 (COX-2), and microsomal PGE synthase-1 (m-PGES-1) [1].
AT-56 inhibits L-PGDS activity in a competitive manner against the substrate PGH₂ with a Kᵢ value of 75 μM. The Kₘ value of L-PGDS for PGH₂ is 14 μM [1]. |
|---|---|
| 体外研究 (In Vitro) |
在表达 L-PGDS 的人类髓母细胞瘤 TE-671 细胞中,AT-56 (1-30 μM; 10 分钟) 以剂量依赖的方式抑制 PGD2 的合成,IC50 约为 3 μM [1]。
AT-56 以浓度 (10-250 μM) 依赖的方式抑制人脑脊液 L-PGDS (β-trace) 和重组小鼠 L-PGDS C89A/C186A 突变体的 PGDS 活性。在 250 μM 浓度下,L-PGDS 活性被抑制至对照组的 30%,IC₅₀ 值约为 95 μM [1]。 浓度高达 250 μM 的 AT-56 对 COX-1、COX-2、m-PGES-1 或 H-PGDS 的活性没有显著影响 [1]。 动力学分析表明,AT-56 以与 PGH₂ 竞争的方式抑制重组 L-PGDS。Vₘₐₓ 保持不变,而 Kₘ 随 AT-56 浓度(0-120 μM)的增加而增加 [1]。 核磁共振滴定分析表明,AT-56 与 L-PGDS 的催化口袋(包含催化中心 Cys⁶⁵)结合,但不与类视黄醇结合口袋结合。 AT-56 结合后,Ser⁵²、Thr⁸⁰、Met⁹⁴ 和 His¹¹⁶ 残基的化学位移发生了显著变化(>0.08 ppm)[1]。 荧光猝灭研究表明,AT-56 结合于 L-PGDS AB 环中 Trp⁵⁴ 附近,但不结合于类视黄醇结合口袋中的 Trp⁴³。在 10 μM AT-56 存在下,荧光强度下降至约 60% [1]。 AT-56 抑制了经 Ca²⁺ 离子载体(5 μM A23187)刺激的表达 L-PGDS 的人 TE-671 细胞中 PGD₂ 的产生,IC₅₀ 值约为 3 μM,但不影响其 PGE₂ 和 PGF₂α 的产生。 AT-56对表达H-PGDS的人类巨核细胞MEG-01S产生PGD₂没有影响[1]。 |
| 体内研究 (In Vivo) |
AT-56(1-30 mg/kg;口服)可降低刺伤脑组织中PGD₂的生成[1]。AT-56(1-10 mg/kg;口服)可抑制小鼠L-PGDS介导的过敏性气道炎症[1]。AT-56(10 mg/kg;口服)的血药浓度峰值(Cmax)为2.15 μg/ml,半衰期为1.71小时,且口服生物利用度极佳(82%)[1]。在H-PGDS基因敲除小鼠(仅表达L-PGDS)中,于刺伤前1小时口服AT-56(1-30 mg/kg)可剂量依赖性地降低脑组织中PGD₂的生成。在 30 mg/kg 剂量下,PGD₂ 水平降至对照组的 40%,而 PGE₂ 和 PGF₂α 水平未受显著影响 [1]。
在卵清蛋白诱导肺部炎症的人 L-PGDS 转基因小鼠中,在抗原暴露前 1 小时和暴露后 24 小时口服 AT-56(1 和 10 mg/kg)可剂量依赖性地降低支气管肺泡灌洗液中的总细胞数、嗜酸性粒细胞数和单核细胞数。在 10 mg/kg 剂量下,总细胞数降至载体对照组的 23%,嗜酸性粒细胞数降至 6%,单核细胞数降至 41% [1]。 |
| 酶活实验 |
以 10 μM [1-¹⁴C]PGH₂ 为底物,在含有 1 mM GSH、0.1 mg/ml IgG 和 10% DMSO 的 100 mM Tris-HCl (pH 8.0) 缓冲液中测定 L-PGDS 活性。反应在 25°C 下孵育 30 秒。产物通过薄层色谱法分离,并使用成像板系统进行定量[1]。
COX-1 和 COX-2 活性测定以 50 μM [1-¹⁴C]花生四烯酸为底物,在含有 2 μM 血红素、5 mM L-色氨酸、0.1 mg/ml IgG 和 10% DMSO 的 100 mM Tris-HCl (pH 8.0) 缓冲液中进行[1]。 动力学分析中,将重组小鼠 L-PGDS C89A/C186A 突变体与不同浓度的 PGH₂ (3-20 μM) 在 100 mM Tris-HCl (pH 8.0) 缓冲液和 1 mM 二硫苏糖醇中孵育,并加入 0、40、100 或 120 μM 的 AT-56(溶于 10% DMSO)。采用 Lineweaver-Burk 图法测定动力学常数[1]。 |
| 细胞实验 |
TE-671细胞(表达L-PGDS的人髓母细胞瘤细胞)和MEG-01S细胞(表达H-PGDS的人巨核细胞)培养于含10% FBS的DMEM培养基中。MEG-01S细胞经TPA诱导分化,使其表达H-PGDS和COX-1。细胞预先用AT-56(0-100 μM)处理10-15分钟,然后在37°C下用5 μM A23187刺激15分钟。收集培养基,并通过酶联免疫吸附试验(ELISA)定量PGD₂、PGE₂和PGF₂α[1]。
对于放射性标记实验,细胞在检测前用[1-¹⁴C]花生四烯酸(3.7 kBq/孔)预标记12小时。刺激后,提取放射性代谢物,通过薄层色谱法分离,并进行放射自显影分析[1]。 |
| 动物实验 |
动物/疾病模型: H-PGDS KO小鼠,脑刺伤(14-16周龄,25-30克,C57BL/6品系)[1]
剂量: 0、1、3、10、30 mg/kg 给药方法:刺伤前1小时口服 实验结果: 抑制脑内L-PGDS反应。使用30 mg/kg AT-56可使脑内PGD2总量减少至40%。 动物/疾病模型: 过表达人 L-PGDS 的 TG 小鼠(雄性,14-16 周,25-30 克)[1] 剂量: 0、1、10 mg/kg 给药途径: 抗原暴露前 1 小时和暴露后 24 小时,每小时口服一次 实验结果: 通过抑制转基因人 L-PGDS 来预防嗜酸性粒细胞浸润。 动物/疾病模型:雄性C57BL/6小鼠(7周龄,22-26克)[1] 剂量:口服10毫克/千克,静脉注射2毫克/千克(药代动力学/PK/PK分析)给药途径:口服和静脉注射 实验结果:口服生物利用度(82%);Cmax(2.15微克/毫升);T1/2(1.71小时),口服); T1/2(2.35 小时),静脉注射。 对于刺伤性脑损伤:H-PGDS KO 小鼠(14-16 周龄,C57BL/6 品系)在损伤前 1 小时口服 AT-56(1、3、10 和 30 mg/kg 体重),该药物溶解于 0.5% 甲基纤维素溶液中。在戊巴比妥麻醉(50 mg/kg)下,将 25 号针头插入额叶皮层(距前囟尾侧 2 mm,距矢状缝外侧 2 mm,深度 1 mm)。脑组织在损伤后10分钟取出,液氮速冻,并保存于-80°C直至进行PG测定[1]。 肺部炎症模型:人L-PGDS转基因小鼠(FVB品系)于第0天和第14天腹腔注射10 μg卵清蛋白(溶于0.2 ml氢氧化铝凝胶)进行致敏。第21天,小鼠暴露于雾化卵清蛋白(50 mg/ml,溶于无菌生理盐水)20分钟。AT-56(1和10 mg/kg)于抗原暴露前1小时和暴露后24小时口服给药。攻击后48小时收集支气管肺泡灌洗液进行细胞计数[1]。 对于刺伤性脑损伤:H-PGDS KO小鼠(14-16周龄,C57BL/6品系)在损伤前1小时口服AT-56(1、3、10和30 mg/kg体重),该药物溶于0.5%甲基纤维素溶液中。在戊巴比妥麻醉(50 mg/kg)下,将25号针头插入额叶皮层(距前囟尾侧2 mm,距矢状缝外侧2 mm,深度1 mm)。脑组织在损伤后10分钟取出,液氮速冻,并保存于-80°C直至进行PG测定[1]。 肺部炎症模型:人L-PGDS转基因小鼠(FVB品系)于第0天和第14天腹腔注射10 μg卵清蛋白(溶于0.2 ml氢氧化铝凝胶)进行致敏。第21天,小鼠暴露于雾化卵清蛋白(50 mg/ml,溶于无菌生理盐水)20分钟。AT-56(1和10 mg/kg)于抗原暴露前1小时和暴露后24小时口服给药。激发后48小时收集支气管肺泡灌洗液进行细胞计数[1]。 |
| 药代性质 (ADME/PK) |
雄性C57BL/6小鼠(7周龄)单次口服10 mg/kg AT-56或单次静脉注射2 mg/kg AT-56。采用高效液相色谱-质谱联用(HPLC-MS)法测定不同时间点的血浆浓度[1]。口服给药后,血浆AT-56浓度在30分钟内(tₘₐₓ = 0.5 h)达到峰值浓度(Cₘₐₓ = 2.15 μg/ml),并随时间推移而下降,12小时后降至检测限以下(0.4 ng/ml)。口服给药的半衰期 (t₁/₂) 为 1.71 小时,静脉给药的半衰期为 2.35 小时 [1]。
静脉注射(2 mg/kg)的浓度-时间曲线下面积 (AUC) 为 1.28 μg·h/ml,口服(10 mg/kg)的浓度-时间曲线下面积为 8.95 μg·h/ml。生物利用度 (BA) 计算为 82%,表明小鼠口服吸收良好 [1]。 |
| 毒性/毒理 (Toxicokinetics/TK) |
口服AT-56后,即使剂量高达100 mg/kg,也未检测到急性毒性作用[1]。
在鸡胚绒毛尿囊膜(CAM)试验中,AT-56剂量高达每枚鸡蛋200 ng时,未引起鸡胚坏死或任何可见的不良反应[1]。 该研究指出,AT-56选择性抑制PGD₂的产生,而不影响其他前列腺素(PGE₂、PGF₂α)的产生,这表明它可能避免因抑制细胞保护性和抗炎性前列腺素而引起的副作用[1]。 |
| 参考文献 | |
| 其他信息 |
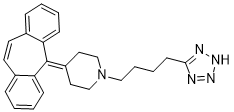
AT-56(4-二苯并[a,d]环庚烯-5-亚基-1-[4-(2H-四唑-5-基)-丁基]-哌啶)是HQL-79(一种H-PGDS抑制剂)的衍生物,已被发现对L-PGDS具有抑制活性[1]。
PGD₂是一种参与睡眠调节和炎症反应的脂质介质。它通过DP₁和DP₂受体发挥作用。L-PGDS促进中枢神经系统、眼部组织、心血管系统和男性生殖器官中PGD₂的生成,并参与睡眠调节、性别决定、动脉粥样硬化保护和脂肪生成[1]。 L-PGDS是脂质运载蛋白家族中唯一的酶。它还能以高亲和力(Kd = 20-200 nM)结合并转运多种亲脂性物质,包括视黄酸、视黄醛、胆绿素、胆红素、神经节苷脂和β-淀粉样蛋白肽,提示其可能作为转运蛋白和内源性分子伴侣发挥作用[1]。 AT-56是首个报道的L-PGDS对PGH₂的竞争性抑制剂。其选择性和口服生物利用度使其成为开发选择性L-PGDS抑制剂的有用原型分子,这些抑制剂可能作为抗嗜睡或抗炎药物发挥作用[1]。 |
| 精确质量 |
397.226
|
|---|---|
| 元素分析 |
C, 75.54; H, 6.85; N, 17.62
|
| CAS号 |
162640-98-4
|
| PubChem CID |
11741525
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
620.4±65.0 °C at 760 mmHg
|
| 闪点 |
329.0±34.3 °C
|
| 蒸汽压 |
0.0±1.8 mmHg at 25°C
|
| 折射率 |
1.646
|
| LogP |
5.84
|
| tPSA |
57.7
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
591
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
LQNGMDUIRLSESZ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C25H27N5/c1-3-9-22-19(7-1)12-13-20-8-2-4-10-23(20)25(22)21-14-17-30(18-15-21)16-6-5-11-24-26-28-29-27-24/h1-4,7-10,12-13H,5-6,11,14-18H2,(H,26,27,28,29)
|
| 化学名 |
1-[4-(2H-tetrazol-5-yl)butyl]-4-(2-tricyclo[9.4.0.03,8]pentadeca-1(15),3,5,7,9,11,13-heptaenylidene)piperidine
|
| 别名 |
AT-56 AT56 AT 56
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~251.56 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.29 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.29 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.29 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00212758 | COMPLETEDWITH RESULTS | Drug: Nutropin AQ | Growth Hormone Deficiency Kidney Failure, Chronic |
Oregon Health and Science University | 2005-01 | Phase 4 |
| NCT01385189 | COMPLETEDWITH RESULTS | Biological: 10 μg Na-GST-1/Alhydrogel Biological: 30 μg Na-GST-1/Alhydrogel Biological: 100 μg Na-GST-1/Alhydrogel |
Hookworm Disease Hookworm Infection |
Baylor College of Medicine | 2012-05 | Phase 1 |
| NCT03296410 | UNKNOWN STATUS | Biological: EV71 and two measles attenuated live vaccine Biological: EV71 and attenuated Japanese encephalitis vaccine Biological: two measles attenuated live vaccine |
Hand, Foot and Mouth Disease (HFMD) | Chinese Academy of Medical Sciences | 2017-09-14 | Phase 4 |
| NCT01261130 | COMPLETED | Biological: 10 μg Na-GST-1/Alhydrogel Biological: 30 μg Na-GST-1/Alhydrogel Biological: 100 μg Na-GST-1/Alhydrogel |
Hookworm Disease Hookworm Infection |
Baylor College of Medicine | 2011-11 | Phase 1 |
| NCT01925417 | COMPLETEDWITH RESULTS | Biological: RBX2660 (microbiota suspension) | Recurrent Clostridium Difficile Infection | Rebiotix Inc | 2013-08 | Phase 2 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
