| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
P2Y12 Receptor ( IC50 = 11 nM )
AZD1283 has an IC50 value of 3.6 μM, demonstrating good antiplatelet aggregation potency[1]. AZD1283 exhibits strong inhibitory effects on CYP450, as evidenced by its IC50 values of 6.62 μM, 0.399 μM, 4.28 μM, and 3.64 μM for CYP2C9, CYP2C19, CYP3A4 (requiring midazolam as substrate), and CYP3A4 (requiring testosterone as substrate), respectively[1]. AZD1283 has an antithrombotic EC50 value of 3 μg/(kg×min) and increases blood flow while inhibiting ADP-induced platelet aggregation[2]. |
|---|---|
| 体外研究 (In Vitro) |
AZD1283 表现出优异的抗血小板聚集效力,IC50 值为 3.6 μM[1]。 AZD1283 对 CYP450 具有高度抑制活性,对 CYP2C9、CYP2C19、CYP3A4(咪达唑仑为底物)和 CYP3A4(睾酮为底物)的 IC50 值分别为 6.62 μM、0.399 μM 和 4.28 μM 和 3.64 μM[1]。 AZD1283 诱导血流量增加并抑制 ADP 诱导的血小板聚集,抗血栓 EC50 值为 3 μg/(kg×min)[2]。
在 ADP 诱导(10 µM)的人富血小板血浆聚集实验中,AZD1283 的 IC50 为 3.60 µM。 [1] 在 CYP450 抑制实验中,AZD1283 对 CYP2C19 显示出强抑制作用(IC50 = 0.399 µM),对 CYP2C9(IC50 = 6.62 µM)和 CYP3A4(以咪达唑仑为底物 IC50 = 4.28 µM,以睾酮为底物 IC50 = 3.64 µM)具有中等抑制作用。 [1] |
| 体内研究 (In Vivo) |
AZD1283 在大鼠(T1/2 = 6.08 分钟)中表现出较差的肝微粒体稳定性,但在狗微粒体(T1/2 = 201 分钟)和人微粒体(T1/2 = 65.0 分钟)中表现出更好的稳定性[1]。动物模型:Sprague-Dawley 大鼠[1] 剂量:5 mg/kg 给药方式:口服;单剂量结果:Cmax 为 25.9 ± 11 ng/mL,T1/2 为 1.68 ± 0.37 h,Tmax 为 0.25 h。
在大鼠氯化铁诱导的动脉血栓形成模型中,口服 10 mg/kg 剂量的 AZD1283 与溶剂对照组相比,血栓重量未观察到显著差异。 [1] |
| 酶活实验 |
体外血小板聚集实验流程:通过 300 g 离心 10 分钟制备人富血小板血浆。通过 2000 g 进一步离心 10 分钟获得贫血小板血浆。使用双通道聚集仪在 37°C 搅拌条件下分析聚集。将富血小板血浆与测试化合物(终浓度:3、10、30、100、300 µM,溶于 0.5% DMSO)或溶剂在 37°C 孵育 2 分钟。随后用 ADP(终浓度 10 µM)刺激血小板。通过记录光密度变化监测聚集 5 分钟。计算抑制率,并通过非线性回归确定 IC50 值。 [1]
CYP450 抑制鸡尾酒法实验流程:实验在 96 孔板中进行。将人肝微粒体、测试化合物或阳性对照抑制剂、以及各 CYP450 亚型的特异性探针底物(CYP1A2 用非那西丁,CYP2C9 用甲苯磺丁脲,CYP2C19 用美芬妥英,CYP2D6 用右美沙芬,CYP3A4 用咪达唑仑和睾酮)在 Tris 缓冲液(pH 7.4)中孵育。37°C 预孵育 10 分钟后,加入 NADPH(终浓度 1 mM)启动反应。37°C 继续孵育 15 分钟后,用含有内标的乙腈淬灭反应。离心后,取上清液进行 LC/MS/MS 分析。在五个不同化合物浓度下测定抑制率,并计算 IC50 值。 [1] |
| 动物实验 |
Sprague-Dawley大鼠
5 mg/kg 口服;单次给药 对于大鼠FeCl3血栓模型,采用雄性Wistar大鼠(250-300 g)。将AZD1283或溶剂悬浮于0.5%羧甲基纤维素钠(CMC-Na)溶液中,并以10 mg/kg(10 mL/kg)的剂量口服给药。给药1.5小时后,对大鼠进行麻醉。分离出一段左侧颈动脉。将浸有20% FeCl3溶液的滤纸置于动脉上10分钟以诱导血栓形成。30分钟后,解剖动脉,取出血栓,清洗、干燥并称重。 [1] 在大鼠药代动力学研究中,使用雄性Sprague-Dawley大鼠(200-220 g)。AZD1283(溶于5% DMSO + 95% HPMC)以5 mg/kg的剂量口服给药。在指定时间点(最长24小时)通过球后静脉采集血样。采用LC/MS/MS法定量分析血浆药物浓度。[1] |
| 药代性质 (ADME/PK) |
在大鼠肝微粒体中,AZD1283 的代谢稳定性极差,消除半衰期 (T1/2) 为 6.08 分钟,固有清除率 (Clint) 为 345 mL/min/g 蛋白,代谢生物利用度 (MF%) 为 11.0%。在犬肝微粒体中,T1/2 为 201 分钟,Clint 为 11.2 mL/min/g 蛋白,MF% 为 66.0%。在人肝微粒体中,T1/2 为 65.0 分钟,Clint 为 32.3 mL/min/g 蛋白,MF% 为 38.0%。 [1]
在雄性 Sprague-Dawley 大鼠单次口服 5 mg/kg 剂量后,AZD1283 显示出较差的药代动力学特性:血浆峰浓度 (Cmax) 为 25.9 ± 11 ng/mL,达峰时间 (Tmax) 为 0.25 h,消除半衰期 (T1/2) 为 1.68 ± 0.37 h,浓度-时间曲线下面积 (AUC0-∞) 为 34.0 ± 3.34 ng·h/mL,平均停留时间 (MRT) 为 2.67 ± 0.77 h。 [1] 在不同基质中测定了AZD1283的溶解度:在肠液中的最大溶解浓度为15.61 ± 6.40 µM,在胃液(pH 1.2)中为0.51 ± 0.20 µM,在水中为2.99 ± 1.80 µM。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
然而,值得注意的是,AZD1283 会抑制多种 CYP450 酶(CYP2C9、CYP3A4 和 CYP2C19),这表明可能存在药物相互作用。[1] 在人醚激活相关基因 (hERG) 检测中,新型优化化合物 58l(源自 AZD1283)在浓度高达 40 µM 时未显示抑制作用,但该数据并非针对 AZD1283 本身。[1]
|
| 参考文献 |
|
| 其他信息 |
AZD1283 是阿斯利康公司报道的一种口服可逆性 P2Y12 受体拮抗剂。它在动物模型中显示出强大的抗血栓疗效和降低出血风险的作用,并进入了人体临床试验。然而,由于吸收不良和酯基代谢稳定性低,其研发在 II 期临床试验前终止。[1] AZD1283 与人 P2Y12 受体结合的 X 射线晶体结构显示,酯羰基与 Asn159 之间的氢键对其高亲和力至关重要。这一发现指导了通过环化酯基来设计稳定性更高的类似物。[1]
|
| 分子式 |
C23H26N4O5S
|
|---|---|
| 分子量 |
470.541344165802
|
| 精确质量 |
470.162
|
| 元素分析 |
C, 58.71; H, 5.57; N, 11.91; O, 17.00; S, 6.81
|
| CAS号 |
919351-41-0
|
| PubChem CID |
23649325
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.4±0.1 g/cm3
|
| 折射率 |
1.618
|
| LogP |
2.66
|
| tPSA |
137.84
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
835
|
| 定义原子立体中心数目 |
0
|
| SMILES |
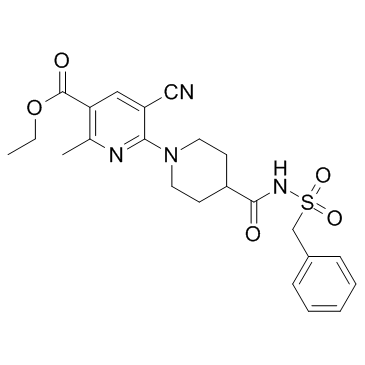
N#CC1C(N2CCC(C(NS(CC3C=CC=CC=3)(=O)=O)=O)CC2)=NC(C)=C(C(OCC)=O)C=1
|
| InChi Key |
NEMHKCNXXRQYRF-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C23H26N4O5S/c1-3-32-23(29)20-13-19(14-24)21(25-16(20)2)27-11-9-18(10-12-27)22(28)26-33(30,31)15-17-7-5-4-6-8-17/h4-8,13,18H,3,9-12,15H2,1-2H3,(H,26,28)
|
| 化学名 |
ethyl 6-[4-(benzylsulfonylcarbamoyl)piperidin-1-yl]-5-cyano-2-methylpyridine-3-carboxylate
|
| 别名 |
AZD-1283; AZD1283; AZD 1283
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~100 mg/mL (~212.5 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.31 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.31 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1252 mL | 10.6261 mL | 21.2522 mL | |
| 5 mM | 0.4250 mL | 2.1252 mL | 4.2504 mL | |
| 10 mM | 0.2125 mL | 1.0626 mL | 2.1252 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Rp-UTP-α-S tetrasodium
Rp-UTP-α-S tetrasodium
 Rp-dUTPαS tetrasodium
Rp-dUTPαS tetrasodium
 MRS2690 disodium
MRS2690 disodium
 P2Y1 antagonist 4
P2Y1 antagonist 4
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA

 463611831
463611831
