| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
PPARδ (EC50 = 1.1 nM)
The target of Endurobol (GW501516; GSK-516) is peroxisome proliferator-activated receptor delta (PPARδ, also known as PPARβ/δ), a nuclear receptor involved in metabolism, inflammation, and cell survival. For human PPARδ: the half-maximal effective concentration (EC₅₀) in luciferase reporter assay is 1.1 nM [5] ; it exhibits high selectivity, with EC₅₀ > 10 μM for PPARα and PPARγ (no significant activation of other PPAR subtypes) [5] . |
|---|---|
| 体外研究 (In Vitro) |
过氧化物体增殖物激活受体β/δ(PPARβ/δ)的激活与抑制癌症细胞系的增殖和凋亡有关。然而,关于PPARβ/δ在鼻咽癌(NPC)中的作用,目前的数据有限。本研究旨在确定PPARβ/δ对人鼻咽癌细胞系中细胞增殖、锚定依赖性克隆形成和异位异种移植物的影响。与对照NP-69细胞相比,低分化和未分化的NPC细胞系中PPARβ/δ的基因和蛋白质表达特异性降低。GW501516(一种特异性PPARβ/δ选择性激动剂)对PPARβ/δ的配体激活显著抑制了细胞增殖和集落形成,并诱导EBV阳性未分化NPC C666-1细胞相对于对照细胞进入G2/M期阻滞。此外,GW501516以半胱天冬酶和BAX依赖的方式诱导C666-1细胞凋亡。根据体外结果,GW501516显著抑制了BALB/cnu/nu小鼠中源自C666-1 NPC细胞的异位NPC异种移植物的致瘤性。这种效应与通过激活AMPKα依赖性信号通路抑制整合素连接激酶(ILK)的基因和蛋白质表达密切相关。总的来说,我们发现PPARβ/δ的表达与鼻咽癌细胞系的分化程度呈负相关,并揭示了GW501516通过激活AMPKα在鼻咽癌细胞中的抗肿瘤作用。这项研究表明,PPARβ/δ靶向分子可能对较差,特别是未分化的鼻咽癌化学预防有用。[5]
GW-501516 是最有效、最具选择性的 PPARα 激动剂,针对 PPARα 的 EC50 为 1.1 nM,选择性是其他人类亚型(PPARα 和-γ)的 1000 倍[1]。 GW 501516 在小鼠培养的近端肾小管 (mProx) 细胞中发挥抗炎作用。 GW 501516 以剂量依赖性方式抑制棕榈酸和 TNFα 诱导的 MCP-1 mRNA 表达增加[3]。 1. PPARδ激活及下游基因调控: - 在转染人类PPARδ表达质粒和PPAR响应元件(PPRE)-荧光素酶报告质粒的HEK293细胞中,0.001–10 μM Endurobol以浓度依赖方式激活PPARδ转录活性,EC₅₀为1.1 nM。1 μM浓度时,PPARδ靶基因(PGC-1α、CD36、CPT1A)的表达上调2.5–4.0倍(实时荧光定量PCR) [5] - 在C2C12肌管细胞中,0.1–1 μM Endurobol使脂肪酸氧化相关基因(CPT1B、ACOX1)和线粒体生物发生基因(PGC-1α、NRF1)的表达上调1.8–3.2倍(Western blot和PCR) [2] 2. 未分化鼻咽癌细胞的抗肿瘤活性: - 在未分化鼻咽癌细胞C666-1中,1–10 μM Endurobol呈浓度依赖性抗增殖作用,IC₅₀为4.3 μM(MTT实验) [5] - 诱导C666-1细胞凋亡:5 μM处理48小时后,早期凋亡细胞(Annexin V⁺/PI⁻)从溶媒组的3.2%增至28.6%,晚期凋亡细胞(Annexin V⁺/PI⁺)从2.1%增至31.4%(流式细胞术)。Western blot显示剪切型caspase-3(3.5倍)、剪切型PARP(2.8倍)和Bax(2.2倍)上调,Bcl-2下调55% [5] - 克隆形成实验:5 μM Endurobol使C666-1细胞的克隆形成效率较溶媒组降低72% [5] 3. 抑制TAK1-NFκB炎症通路: - 在TNFα(10 ng/mL)刺激的小鼠肾近端小管上皮细胞(mRPTCs)中,0.1–1 μM Endurobol剂量依赖性抑制TAK1(Ser412)和NFκB p65(Ser536)的磷酸化(抑制率40–65%,Western blot)。1 μM浓度时,促炎细胞因子IL-6(降低52%)和TNFα(降低48%)的分泌减少(ELISA) [3] |
| 体内研究 (In Vivo) |
过氧化物酶体增殖物激活受体(PPARs)是配体诱导型转录因子的核受体家族,有三种不同的亚型:PPARα、δ和γ。已经证明PPARα和γ激动剂在蛋白尿性肾病中具有肾脏保护作用;然而,PPARδ激动剂在肾脏疾病中的作用尚不清楚。因此,我们在蛋白质超载小鼠肾病模型中研究了PPARδ激动剂GW501516的肾脏保护作用,并确定了其分子机制。喂食对照饮食或含GW501516饮食的小鼠腹腔注射游离脂肪酸(FFA)结合的白蛋白或PBS(-)。在对照组中,蛋白质超载导致肾小管损伤、巨噬细胞浸润和MCP-1和TNFαmRNA表达增加。GW501516治疗可以预防这些影响。在蛋白尿性肾病中,近端肾小管细胞过度暴露于白蛋白、与白蛋白结合的FFA或TNFα等细胞因子是有害的。使用培养的近端肾小管细胞的体外研究表明,GW501516通过直接抑制TGF-β活化激酶1(TAK1)-NFκB通路(TNFα受体和toll样受体-4的常见下游信号通路)来减弱TNFα和FFA(棕榈酸酯)诱导的MCP-1表达,但不减弱白蛋白诱导的MCP.1表达。总之,我们证明GW501516对肾小管细胞具有抗炎作用,并可作为减轻蛋白尿性肾脏疾病中小管间质病变的治疗候选药物。[3]
运动可以增加骨骼肌中过氧化物酶体增殖物激活受体δ(PPARδ)的表达。PPARδ调节肌肉代谢并重新编程肌肉纤维类型,以提高跑步耐力。本研究利用代谢组学分析来研究PPARδ激动剂GW501516对小鼠跑步耐力的影响。虽然单独训练可以提高力竭性跑步表现,但GW501516治疗提高了训练和未训练小鼠的跑步耐力和琥珀酸脱氢酶(SDH)阳性肌纤维的比例。此外,在训练和/或治疗后,观察到脂肪酸氧化途径中的中间代谢物和关键酶水平升高。单独训练可增加血清肌醇、糖原氨基酸和支链氨基酸。然而,GW501516增加了血清半乳糖和β-羟基丁酸,与训练无关。此外,单独使用GW501516可以提高血清不饱和脂肪酸水平,尤其是多不饱和脂肪酸酯,但与训练相结合时,水平会增加得更多。这些发现表明,GW501516和训练提高跑步能力的机制并不完全相同。训练通过促进蛋白质的分解代谢和糖异生来提高能量可用性,而GW501516则提高了脂肪酸的特定消耗并减少了葡萄糖的利用。[4] 过氧化物体增殖物激活受体β/δ(PPARβ/δ)的激活与抑制癌症细胞系的增殖和凋亡有关。然而,关于PPARβ/δ在鼻咽癌(NPC)中的作用,目前的数据有限。本研究旨在确定PPARβ/δ对人鼻咽癌细胞系中细胞增殖、锚定依赖性克隆形成和异位异种移植物的影响。与对照NP-69细胞相比,低分化和未分化的NPC细胞系中PPARβ/δ的基因和蛋白质表达特异性降低。GW501516(一种特异性PPARβ/δ选择性激动剂)对PPARβ/δ的配体激活显著抑制了细胞增殖和集落形成,并诱导EBV阳性未分化NPC C666-1细胞相对于对照细胞进入G2/M期阻滞。此外,GW501516以半胱天冬酶和BAX依赖的方式诱导C666-1细胞凋亡。根据体外结果,GW501516显著抑制了BALB/cnu/nu小鼠中源自C666-1 NPC细胞的异位NPC异种移植物的致瘤性。这种效应与通过激活AMPKα依赖性信号通路抑制整合素连接激酶(ILK)的基因和蛋白质表达密切相关。总的来说,我们发现PPARβ/δ的表达与鼻咽癌细胞系的分化程度呈负相关,并揭示了GW501516通过激活AMPKα在鼻咽癌细胞中的抗肿瘤作用。这项研究表明,PPARβ/δ靶向分子可能对较差,特别是未分化的鼻咽癌化学预防有用。[5] GW-501516 损害骨形成,导致 OVX 大鼠 BMD 降低和骨特性恶化[2]。 GW 501516 可减轻蛋白质超载小鼠肾病模型中的间质炎症和近端肾小管细胞损伤[3]。 GW 501516 治疗可增强经过训练和未经训练的小鼠的跑步耐力和琥珀酸脱氢酶 (SDH) 阳性肌纤维的比例[4]。 1. 卵巢切除(OVX)大鼠的骨和肌肉影响: - 12周龄雌性Sprague-Dawley大鼠行卵巢切除术,Endurobol(0.3、1、3 mg/kg/天)口服灌胃给药8周。 - 骨组织:1 mg/kg和3 mg/kg剂量使腰椎骨密度(BMD)较OVX溶媒组分别增加12%和18%;股骨BMD分别增加10%和15% [2] - 肌肉组织:3 mg/kg剂量使腓肠肌重量增加14%,比目鱼肌重量增加11%;腓肠肌肌纤维横截面积增加16% [2] - 各剂量对体重或脂肪量无显著影响 [2] 2. 蛋白尿肾病小鼠的肾小管间质炎症改善: - BALB/c小鼠经尾静脉注射阿霉素(10 mg/kg)诱导蛋白尿肾病,Endurobol(3 mg/kg)腹腔注射,隔天1次,持续4周。 - 肾功能:尿蛋白/肌酐比值降低45%,血尿素氮(BUN)降低32%(较溶媒组) [3] - 炎症反应:肾小管间质炎症细胞浸润减少(CD45⁺细胞减少58%);肾组织IL-6和TNFα的mRNA水平分别降低50%和46%(PCR);p-TAK1和p-NFκB p65蛋白水平分别降低62%和55%(Western blot) [3] 3. 昆明小鼠跑步耐力提升: - 雄性昆明小鼠分为对照组和Endurobol(2 mg/kg/天)组,口服灌胃4周,两组均进行跑步机训练(每天30分钟,每周5天)。 - 跑步耐力:力竭时间从对照组的182±15分钟延长至处理组的268±22分钟,提升47% [4] - 代谢变化:肝和腓肠肌中脂肪酸氧化酶CPT1A的水平分别增加2.1倍和1.8倍(Western blot);血浆游离脂肪酸水平降低30% [4] |
| 酶活实验 |
1. PPARδ激活荧光素酶报告基因实验:
- HEK293细胞以4×10⁴个/孔接种于24孔板,用含10%胎牛血清的DMEM培养基孵育过夜 [5] - 采用转染试剂将人类PPARδ表达质粒、PPRE-荧光素酶报告质粒及海肾荧光素酶质粒(内参)共转染至细胞 [5] - 转染24小时后,加入系列浓度的Endurobol(0.001–10 μM)或溶媒(DMSO),继续孵育24小时 [5] - 制备细胞裂解液,采用双荧光素酶报告基因检测系统测定荧光素酶活性,计算萤火虫荧光素酶/海肾荧光素酶的相对活性,从剂量-反应曲线推导EC₅₀值 [5] 2. PPAR亚型选择性实验: - 实验流程与PPARδ激活实验一致,仅将人类PPARδ表达质粒替换为PPARα或PPARγ表达质粒。Endurobol的测试浓度高达10 μM,以评估对PPARδ的选择性 [5] |
| 细胞实验 |
将GW 501516溶解在DMSO中。将细胞在 0.2% FCS DMEM 中孵育 9 小时,然后与终浓度为 2.5 和 5 µM 的 GW 501516 或作为对照的 0.05% DMSO 预孵育 3 小时,然后用 150 µM 棕榈酸酯结合物刺激至 8.0% BSA 12 小时[3]。
1. 鼻咽癌细胞增殖及凋亡实验: - C666-1细胞以5×10³个/孔接种于96孔板(MTT实验)或2×10⁵个/孔接种于6孔板(凋亡实验),用含10%胎牛血清的RPMI 1640培养基孵育过夜 [5] - 增殖实验:加入系列浓度的Endurobol(0.1–20 μM),细胞孵育72小时后加入MTT溶液,570 nm处测定吸光度并计算IC₅₀ [5] - 凋亡实验:用1–10 μM Endurobol处理细胞48小时,收集细胞后用Annexin V-FITC和PI染色,流式细胞术分析 [5] 2. 肾上皮细胞炎症通路实验: - 小鼠肾近端小管上皮细胞(mRPTCs)以1×10⁶个/孔接种于6孔板,孵育过夜。用0.1–1 μM Endurobol预处理细胞1小时,随后用10 ng/mL TNFα刺激6小时 [3] - 用含蛋白酶/磷酸酶抑制剂的RIPA缓冲液裂解细胞,蛋白裂解液用于Western blot分析p-TAK1(Ser412)、总TAK1、p-NFκB p65(Ser536)和总NFκB p65 [3] - 收集培养上清液,通过ELISA测定IL-6和TNFα的浓度 [3] 3. 肌管代谢基因表达实验: - C2C12成肌细胞接种于6孔板,更换为含2%马血清的培养基培养5天分化为肌管细胞。用0.1–1 μM Endurobol处理肌管细胞24小时 [2] - 提取总RNA并合成cDNA,采用实时荧光定量PCR检测PGC-1α、CPT1B和ACOX1的mRNA水平(以GAPDH为内参基因) [2] |
| 动物实验 |
大鼠:12周龄的雌性Sprague Dawley大鼠被分为三组:对照组(OVX-CTR)、低剂量GW 501516组(OVX-GW1)和高剂量GW 501516组(OVX-GW5)。在接下来的四个月里,每天通过灌胃给予动物GW 501516或溶剂(甲基纤维素)。采用双能X射线吸收法评估股骨、脊柱和全身的骨密度(BMD)[2]。
小鼠:小鼠接受治疗性饮食和治疗,并随机分组。为了制备含有GW 501516的啮齿动物饲料,将GW 501516逐渐添加到对照饲料中,直至其最终浓度达到0.04% (w/w)。对照组饮食中10%的热量来源于脂肪(5.5%来自大豆油,4.5%来自猪油)[3]。雌激素缺乏会促进骨质流失和骨骼肌功能障碍。过氧化物酶体增殖物激活受体(PPARs)有3个亚型(α、δ和γ)。PPARγ激动剂会诱导骨质流失,而PPARα激动剂会增加骨量。虽然已知PPARδ激动剂会影响骨骼肌代谢,但其对骨骼的影响尚不明确。本研究探讨了PPARδ激动剂GW501516对卵巢切除(OVX)大鼠肌肉骨骼的影响。12周龄的雌性Sprague Dawley大鼠被随机分为假手术组和3个OVX组;分别设置高剂量 GW501516 组(OVX-GW5)、低剂量 GW501516 组(OVX-GW1)和对照组(OVX-CTR)(每组 n = 12)。动物每日灌胃给予 GW501516 或赋形剂(甲基纤维素),持续 4 个月。采用双能 X 射线吸收法评估股骨、脊柱和全身的骨密度 (BMD)。采用微型计算机断层扫描评估近端胫骨的骨微结构,并进行动态组织形态计量学分析。分析股四头肌形态和线粒体蛋白的相对表达。检测血浆中的骨代谢标志物和代谢标志物。4 个月后,OVX-GW5 组的股骨 BMD 低于 OVX-CTR 组。与 OVX-CTR 组相比,GW501516 治疗组的骨小梁间距更大。与OVX-CTR组相比,OVX-GW5组的皮质面积分数较低,结构模型指数较高。这些效应与两个GW组的骨形成受损相一致。OVX-GW5组的甘油三酯水平升高,脂联素水平降低,但未观察到对肌肉形态或线粒体基因表达的影响。总之,PPARδ激动剂GW501516对OVX大鼠的骨骼特性有负面影响,但未检测到对骨骼肌的影响。[2] 1. 卵巢切除(OVX)大鼠骨骼和肌肉研究模型: -12周龄、体重220-250克的雌性Sprague-Dawley大鼠麻醉后切除双侧卵巢(OVX组);假手术组大鼠作为正常对照。卵巢切除(OVX)大鼠恢复1周后,随机分为4组(每组n=8):OVX载体组(0.5%羧甲基纤维素钠)、Endurobol组(0.3 mg/kg、1 mg/kg、3 mg/kg)[2] - Endurobol悬浮于0.5%羧甲基纤维素钠溶液中,每日灌胃一次,持续8周。假手术组和OVX载体组灌胃相同体积的载体[2] - 治疗结束后,处死大鼠。收集腰椎和股骨进行骨密度(BMD)测量;称量腓肠肌和比目鱼肌的重量,并用苏木精-伊红染色分析肌纤维横截面积[2] 2.蛋白尿肾病小鼠模型: - 将雌性BALB/c小鼠(6-8周龄,18-22 g)随机分为正常对照组、疾病载体组和Endurobol组(每组n=6)。通过尾静脉注射阿霉素(10 mg/kg)诱导疾病模型[3] - 阿霉素注射7天后,将Endurobol(3 mg/kg)溶于5% DMSO + 95%生理盐水中,每隔一天腹腔注射一次,持续4周;疾病载体组注射等体积的DMSO-生理盐水混合液[3] - 第4周时,将小鼠置于代谢笼中收集24小时尿液。处死小鼠后,采集血液样本进行BUN和肌酐检测;采集肾脏进行组织病理学分析、蛋白质印迹和PCR[3] 3.昆明小鼠跑步耐力模型: -雄性昆明小鼠(6-8周龄,20-25 g)随机分为对照组和Endurobol组(每组n=10)。Endurobol(2 mg/kg/天)溶于DMSO(1%)+玉米油(99%)中,每日一次灌胃给药,持续4周;对照组接受DMSO-玉米油混合物[4] - 两组均在为期4周的治疗期间进行跑步机训练(速度:15米/分钟,坡度:5°,每天30分钟,每周5天)[4] - 治疗结束后,在跑步机上测试跑步耐力(起始速度15米/分钟,每3分钟增加1米/分钟直至力竭),并记录力竭时间。采集肝脏和腓肠肌进行Western blot分析[4] |
| 药代性质 (ADME/PK) |
1. 血浆蛋白结合率:通过平衡透析法测定,Endurobol 具有较高的血浆蛋白结合率 (98.5%),且结合率与浓度 (0.1–10 μM) 无关 [5]
2. 口服生物利用度:在大鼠中,口服 Endurobol (3 mg/kg) 的口服生物利用度 (F) 为 72% [2] 3. 末端半衰期:在大鼠中,口服 Endurobol (3 mg/kg) 后,血浆末端半衰期 (t₁/₂) 为 8.3 小时 [2] 4. 组织分布:在小鼠中,Endurobol 主要分布于肝脏、骨骼肌和脂肪组织,2 小时后,组织/血浆浓度比分别为 3.2(肝脏)、2.8(腓肠肌)和 2.5(脂肪组织)。口服后给药(2 mg/kg)[4] |
| 毒性/毒理 (Toxicokinetics/TK) |
1. 体外细胞毒性:浓度高达 20 μM 的 Endurobol 对正常人鼻咽上皮细胞 (NP69) 或正常肾上皮细胞无显著细胞毒性,细胞存活率 >90%(与溶剂对照组相比,MTT 法)[5]
2. 体内亚慢性毒性: - 大鼠接受 Endurobol 治疗(3 mg/kg/天,持续 8 周)后,体重、食物摄入量或血清生化指标(ALT、AST、BUN、肌酐)均无显著变化[2] - 小鼠接受 Endurobol 治疗(隔日 3 mg/kg,持续 4 周)或(2 mg/kg/天,持续 4 周)后,肝脏、肾脏、心脏或肺脏均未出现组织病理学异常[3,4] 3. 无急性毒性:单次口服给药在小鼠中,剂量高达 100 mg/kg 的 Endurobol 在 7 天内不会导致死亡或明显的毒性反应(例如抽搐、嗜睡)[4] |
| 参考文献 |
|
| 其他信息 |
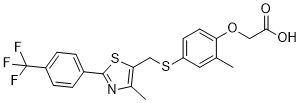
GW 501516 是一种芳香醚,其结构为苯氧乙酸,其中苯基在 2 位被甲基取代,4 位被 (1,3-噻唑-5-基甲基)硫代二基取代,1,3-噻唑基在 2 位和 4 位分别被对三氟甲基苯基和甲基取代。它是一种过氧化物酶体增殖物激活受体 β/δ 激动剂和致癌物。它是一种单羧酸,属于 1,3-噻唑类化合物、有机氟化合物、芳基硫醚和芳香醚。
卡达林 (GW-501516) 是一种过氧化物酶体增殖物激活受体 δ 激动剂,可用于治疗血脂异常。卡达林已被研究用于治疗肥胖症、血脂异常和心血管疾病。 药物适应症 已研究用于治疗高脂血症。 作用机制 该药物调节多种组织(例如骨骼肌和脂肪组织)中的脂肪酸氧化。使用转基因小鼠模型过表达PPARδ可促进肌肉氧化能力的增强。它在以饱和脂肪过量为特征的西方饮食的代谢适应中也发挥着重要作用。 1. Endurobol(GW501516;GSK-516)是一种强效、选择性的小分子PPARδ激动剂,最初是作为药理学研究工具开发的,用于研究PPARδ介导的生物学过程[1] 2. 作用机制:Endurobol与PPARδ的配体结合域结合,诱导构象变化,促进其与RXR形成异二聚体,并与靶基因中的PPRE结合。这激活了参与脂肪酸氧化、线粒体生物合成、肌肉合成代谢、骨代谢和抑制炎症信号(TAK1-NFκB通路)的下游通路。在癌细胞中,它通过调节 Bcl-2/Bax/caspase 级联反应促进细胞凋亡[2,3,4,5] 3. 化学类别:它属于噻唑烷二酮类化合物,分子量为 453.5 g/mol。文献[1]报道了一种简短高效的合成路线。 4. 治疗潜力:基于临床前数据,该化合物在以下方面具有潜在应用价值: - 骨质疏松症和肌肉萎缩(改善卵巢切除大鼠的骨密度和肌肉量)[2] - 蛋白尿性肾病(改善肾小管间质炎症)[3] - 代谢紊乱(增强脂肪酸氧化和运动耐力)[4] - 未分化型鼻咽癌(抑制增殖并诱导细胞凋亡)[5] 5. 研究应用:该化合物被广泛用作研究PPARδ在代谢、炎症、骨/肌肉生理和癌症中功能的工具化合物,有助于验证PPARδ作为治疗靶点的有效性[1-5] |
| 分子式 |
C21H18F3NO3S2
|
|---|---|
| 分子量 |
453.497733592987
|
| 精确质量 |
453.068
|
| 元素分析 |
C, 55.62; H, 4.00; F, 12.57; N, 3.09; O, 10.58; S, 14.14
|
| CAS号 |
317318-70-0
|
| 相关CAS号 |
317318-70-0
|
| PubChem CID |
9803963
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
584.5±60.0 °C at 760 mmHg
|
| 熔点 |
134-136°C
|
| 闪点 |
307.3±32.9 °C
|
| 蒸汽压 |
0.0±1.7 mmHg at 25°C
|
| 折射率 |
1.619
|
| LogP |
6.29
|
| tPSA |
112.96
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
572
|
| 定义原子立体中心数目 |
0
|
| SMILES |
OC(COC1=CC=C(C=C1C)SCC2=C(N=C(S2)C3=CC=C(C=C3)C(F)(F)F)C)=O
|
| InChi Key |
YDBLKRPLXZNVNB-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H18F3NO3S2/c1-12-9-16(7-8-17(12)28-10-19(26)27)29-11-18-13(2)25-20(30-18)14-3-5-15(6-4-14)21(22,23)24/h3-9H,10-11H2,1-2H3,(H,26,27)
|
| 化学名 |
2-[2-methyl-4-[[4-methyl-2-[4-(trifluoromethyl)phenyl]-1,3-thiazol-5-yl]methylsulfanyl]phenoxy]acetic acid
|
| 别名 |
Endurobol; GSK516; GW1516; GW501516; GW 1516; GSK 516; GW-501516; GW-1516; GSK-516; Cardarine; GW 501516
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~51 mg/mL (~197.5 mM)
Water: ~20 mg/mL (~77.5 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.51 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (5.51 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.51 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 2% DMSO+40% PEG 300+2% Tween 80+ddH2O: 6mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2051 mL | 11.0254 mL | 22.0507 mL | |
| 5 mM | 0.4410 mL | 2.2051 mL | 4.4101 mL | |
| 10 mM | 0.2205 mL | 1.1025 mL | 2.2051 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00158899 | Completed | Drug: GW501516 oral tablets | Dyslipidaemias Dyslipidaemia |
GlaxoSmithKline | August 2004 | Phase 2 |
| NCT00388180 | Completed | Drug: GW501516 Drug: GW590735 |
Dyslipidaemias Obesity |
GlaxoSmithKline | December 2004 | |
| NCT00841217 | Completed | Drug: GW501516 Drug: placebo pill |
Obesity Lipid Disorders |
The University of Western Australia |
April 2003 | Phase 4 |
| NCT00318617 | Terminated | Drug: GW510516X | Dyslipidaemias Heart Failure |
GlaxoSmithKline | December 2005 |
 The impact of GW501516 on cell proliferation and colony formation.
GW501516 inhibited gene and protein expression of integrin-linked kinase (ILK) in C666-1 cells.Front Pharmacol.2018 Jun 28;9:648. |
|---|
 The impact of GW501516 on cell cycle of C666-1 cells.
GW501516 suppressed SS tumor growthin vivo.Front Pharmacol.2018 Jun 28;9:648. |
 The impact of GW501516 on apoptosis in C666-1 cells.
The impact of GW501516 on protein expression in C666-1 cells. The gene and protein expression of PPARβ/δ in nasopharyngeal carcinoma (NPC) cell lines.Front |
 VSP-77
VSP-77
 NC-2100
NC-2100
 PPARγ agonist 20
PPARγ agonist 20
 KRP-105
KRP-105
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA




 463611831
463611831
