| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
ANXA7 GTPase; AMPK/mTORC1/STAT3
|
|---|---|
| 体外研究 (In Vitro) |
SEC (20 µM) 可防止 PC3 前列腺癌细胞和 HEK 293T RKIP−/− 细胞迁移 [1]。 ANXA7 GTPase 特异性抑制剂 ABO 可逆转 SEC (20 µM) 引起的 PC3 细胞中 AMPK 磷酸化的显着增加,表明磷酸化水平增加的激活的 ANXA7 可促进 AMPK 磷酸化(细胞选择性)[1]。
SEC通过促进ITGB4核易位特异性诱导ITGB4高表达的肿瘤细胞凋亡。[2] |
| 体内研究 (In Vivo) |
在 PC-3M-Luc 原位植入裸鼠模型中,SEC(3 mg/kg/天或 18 mg/kg/天)可预防转移 [1]。
SEC抑制PC-3M-Luc原位植入裸鼠模型的转移[1]
接下来,我们通过监测小鼠原位前列腺癌转移来评估SEC的体内疗效。原位接种PC-3M-Luc的小鼠分别注射SEC (3 mg/kg/d或18 mg/kg/d) 3周。重要的是,SEC降低了以光子计数表达的生物发光信号(图7,A和B)。淋巴结是前列腺癌的常见转移部位。然后分离主动脉淋巴结,进行体外生物发光成像。PC-3M-Luc细胞能够扩散到远处的器官,这表明存在淋巴结转移。值得注意的是,SEC降低了转移淋巴结的生物发光信号,减少了前列腺癌转移的主动脉淋巴结的数量(图7,C至E)。同时,SEC治疗不影响小鼠的体重(图7F)。一致地,SEC注射确实降低了移植肿瘤中CCL2、APLN和IL6ST的mRNA水平(图8)。这些体内观察表明,SEC抑制了裸鼠模型中PC-3M-Luc细胞的淋巴结转移能力[1]。 SEC抑制人肿瘤异种移植物在禽胚模型中的生长[2] 接下来,我们在体内确定SEC是否具有促进细胞凋亡的作用。鸡胚绒毛尿囊膜(chorioallantoic membrane, CAM)由于其免疫缺陷的环境和致密的毛细血管网络,被广泛用于肿瘤植入,以评估抗癌药物的疗效和促血管生成或抗血管生成因子的作用。因此,我们采用CAM模型来研究SEC在肿瘤生长和正常血管生成中的作用。A549细胞沉积在CAM上,2天内形成实体瘤。从第2天到第8天,用PBS或每2天含SEC的PBS局部治疗实体腺癌。与单独使用PBS相比,PBS加SEC治疗可显著抑制肿瘤生长(图6A)。5FU处理有利于减小异种移植物的大小(补充图6)。肿瘤冷冻切片的TUNEL检测显示,SEC在体内有效促进了细胞凋亡,并且在肿瘤周围边缘检测到更强的信号(图6B)。为了确定SEC是否干扰正常血管生成并抑制肿瘤生长,我们直接测量了SEC对CAM的血管生成作用。SEC对CAM正常血管生成无影响(图6C)。与体内血管生成研究完全一致,在体外Matrigel实验中,SEC没有干扰血清和FGF-2的毛细血管样管结构的形成(图6D)。因此,SEC在体内通过诱导细胞凋亡有效抑制肿瘤生长,对正常CAM血管生成无不良影响。 |
| 酶活实验 |
体外ANXA7活性测定[2]
将人宽型ANXA7、ANXA7-mt1 (T275A)和-mt2 (T286A)突变体的编码区亚克隆到带6*His标签的mCherry-N1表达载体中。当HEK293在10 cm培养皿上的密度达到70% ~ 80%合流时,用指定的表达载体转染细胞。横切24 h后,收获总蛋白,用His gravrap Ni-NTA蛋白纯化试剂盒提取表达的重组蛋白ANXA7并进行柱层析纯化。纯化的ANXA7在37°C下加或不加SEC孵育,孵育指定的浓度和时间。使用atp酶/GTPase活性测定试剂盒测定ANXA7的GTPase活性。 |
| 细胞实验 |
细胞活力测定[1]
细胞类型: HEK293T RKIP−/− 细胞。 [1] 测试浓度: 20 µM。 孵化持续时间:24 小时。 实验结果:抑制HEK293T RKIP−/−细胞的迁移,但对HEK293T RKIP+/+细胞无影响。 |
| 动物实验 |
动物/疾病模型:将荧光素酶标记的PC-3M-Luc细胞(2 × 10⁶个细胞/50 µL无菌HBSS−/−)原位接种到8周龄裸鼠的前列腺中[1]。
剂量:3 mg/kg/天或18 mg/kg/天。 给药方式:每日腹腔注射,持续3周。 实验结果:抑制裸鼠模型中PC-3M-Luc细胞的淋巴结转移能力。降低移植肿瘤中CCL2、APLN和IL6ST的mRNA水平。不影响小鼠体重。 |
| 参考文献 |
|
| 其他信息 |
Annexin A7 (ANXA7) 是前列腺癌肿瘤发生和转移的抑制因子。活化的 ANXA7 GTPase 可促进前列腺癌细胞凋亡。然而,ANXA7 GTPase 在前列腺癌转移中的作用及其潜在机制尚未明确。RKIP 是一种转移抑制因子,在前列腺癌转移灶中表达下调。RKIP 与其靶蛋白的结合可能抑制其相互作用蛋白的活化。然而,RKIP 对 ANXA7 GTPase 活化的影响尚不清楚。本研究发现,小分子 SEC((S)-1-(3-(4-氯苯氧基)-2-羟丙基)-3-(4-甲氧基苯基)-1H-吡唑-5-羧酸乙酯)可有效激活 ANXA7 GTPase,从而抑制前列腺癌转移。从机制上讲,活化的ANXA7促进AMPK磷酸化,导致mTORC1活性降低,STAT3核转位受到抑制,以及包括CCL2、APLN和IL6ST在内的促转移基因表达下调。相反,RKIP与ANXA7相互作用,并抑制SEC及其下游信号通路对ANXA7 GTP酶的激活。值得注意的是,在体内原位分析中,SEC处理抑制了前列腺癌细胞的转移。总之,我们的研究结果为低RKIP表达的前列腺癌转移如何通过SEC诱导的AMPK/mTORC1/STAT3信号通路激活ANXA7 GTP酶而受到抑制提供了新的见解。[1]整合素β4 (ITGB4)水平升高与多种癌的恶性进展相关。然而,目前尚未有针对高表达ITGB4的癌细胞的选择性治疗策略报道。本文首次报道了一种手性小分子SEC,它通过诱导ITGB4核转位,选择性地促进高表达ITGB4的癌细胞凋亡。核内ITGB4可与ATF3启动子区域结合,激活ATF3的表达,进而上调下游促凋亡基因的表达。此外,SEC促进膜联蛋白A7 (ANXA7)与ITGB4的结合,并增强ANXA7的GTP酶活性。活化的ANXA7通过触发ITGB4在Y1494位点的磷酸化,促进ITGB4的核转位。SEC还能抑制禽类胚胎模型中异种移植瘤的生长。我们发现了一种小分子SEC,它通过触发ITGB4与ANXA7的结合、ITGB4的核转位以及促凋亡基因的表达,在体外和体内均对高表达ITGB4的癌细胞具有选择性促凋亡作用。[2]
|
| 分子式 |
C22H23CLN2O5
|
|---|---|
| 分子量 |
430.881425142288
|
| 精确质量 |
430.1295
|
| 元素分析 |
C, 61.33; H, 5.38; Cl, 8.23; N, 6.50; O, 18.57
|
| CAS号 |
1802997-81-4
|
| 相关CAS号 |
1802997-81-4;918879-70-6 (recemate);
|
| PubChem CID |
146681186
|
| 外观&性状 |
Light yellow to yellow ointment
|
| LogP |
4
|
| tPSA |
82.8Ų
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
10
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
524
|
| 定义原子立体中心数目 |
1
|
| SMILES |
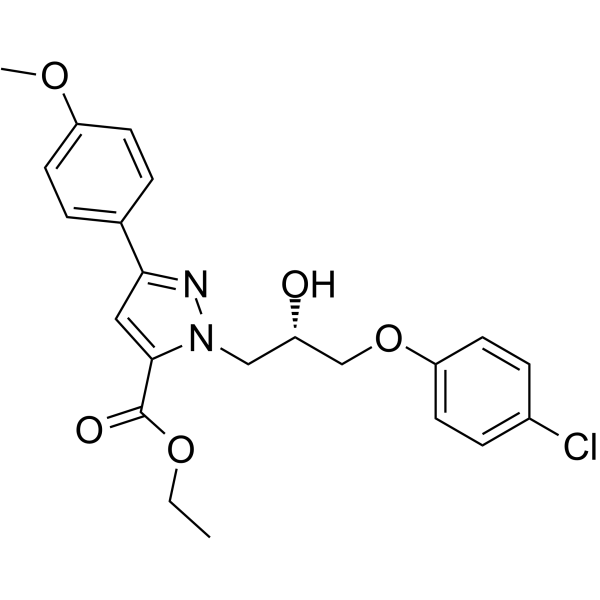
O=C(C1=CC(C2=CC=C(OC)C=C2)=NN1C[C@H](O)COC3=CC=C(Cl)C=C3)OCC
|
| InChi Key |
OFKGGSVHXHMDDN-KRWDZBQOSA-N
|
| InChi Code |
InChI=1S/C22H23ClN2O5/c1-3-29-22(27)21-12-20(15-4-8-18(28-2)9-5-15)24-25(21)13-17(26)14-30-19-10-6-16(23)7-11-19/h4-12,17,26H,3,13-14H2,1-2H3/t17-/m0/s1
|
| 化学名 |
ethyl 2-[(2S)-3-(4-chlorophenoxy)-2-hydroxypropyl]-5-(4-methoxyphenyl)pyrazole-3-carboxylate
|
| 别名 |
Sec; 1802997-81-4; ethyl 2-[(2S)-3-(4-chlorophenoxy)-2-hydroxypropyl]-5-(4-methoxyphenyl)pyrazole-3-carboxylate;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~232.08 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.80 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (5.80 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.80 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3208 mL | 11.6042 mL | 23.2083 mL | |
| 5 mM | 0.4642 mL | 2.3208 mL | 4.6417 mL | |
| 10 mM | 0.2321 mL | 1.1604 mL | 2.3208 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
