| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
JAK3-IN-6 (Compound 2) is a powerful JAK3 inhibitor with a concentration of 0.15 nM. In an enzymatic assay, it demonstrates a 4300-fold greater selectivity for JAK3 than JAK1, meaning that it is 67-fold (IL-2 vs. IL-6) or 140-fold (IL-2 vs. EPO or GMCSF) selective in cell reporter gene assays and >35-fold (IL-7 vs. IL-6 or GMCSF) in human PBMC assays. In order to ascertain whether there is an additive or synergistic effect of synergy, JAK3-IN-6 was cross-titrated with JAK1 selective inhibitor 3 (JAK1: 0.96 nM, JAK2: 14 nM, JAK3: >1500 nM, TYK2: 10 nM). This cross-titration was irreversible. shows that the predicted pSTAT5 inhibition levels based on the addition of JAK1 and JAK3 inhibition are very close to the measured effects of cross-titration of each compound, indicating that inhibition of JAK1 and JAK3 is Blocking STAT5 phosphorylation has additive but not synergistic effects. Inhibits the JAK1 and JAK3 enzymes that inhibit IL-7 signaling in CD3+, CD4+ PBMC. Furthermore, JAK1 or JAK3 inhibition on its own is adequate to totally block pSTAT5 [2].
|
|---|---|
| 体外研究 (In Vitro) |
JAK3-IN-6(化合物 2)是一种强效 JAK3 抑制剂,浓度为 0.15 nM。在酶测定中,它对 JAK3 的选择性比 JAK1 高 4300 倍,这意味着它的选择性是 67 倍(IL-2 与 IL-6)或 140 倍(IL-2 与 EPO 或 GMCSF)在细胞报告基因测定中,在人 PBMC 测定中超过 35 倍(IL-7 与 IL-6 或 GMCSF)。为了确定是否存在协同作用的累加或协同效应,将 JAK3-IN-6 与 JAK1 选择性抑制剂 3 进行交叉滴定(JAK1:0.96 nM,JAK2:14 nM,JAK3:>1500 nM,TYK2:10 nM )。这种交叉滴定是不可逆的。显示基于添加JAK1和JAK3抑制的预测的pSTAT5抑制水平非常接近每种化合物的交叉滴定的测量效果,表明JAK1和JAK3的抑制与阻断STAT5磷酸化具有相加作用,但没有协同作用。抑制 JAK1 和 JAK3 酶,从而抑制 CD3+、CD4+ PBMC 中的 IL-7 信号传导。此外,JAK1 或 JAK3 抑制本身足以完全阻断 pSTAT5 [2]。
在单JAK亚型PathHunter细胞实验(经改造通过特定JAK传导信号)中,JAK3-IN-6 抑制JAK2/3嵌合体信号传导的IC50为380 nM,对JAK1 (IC50 = 50,000 nM) 的选择性为80倍,对JAK2 (IC50 >50,000 nM) 的选择性超过130倍。[2] 在细胞因子报告基因(Cellsensor)实验中,JAK3-IN-6 抑制IL-2 (JAK1/JAK3) 信号传导的IC50为70 nM。与IL-6 (JAK1/JAK2) 信号传导 (IC50 = 4710 nM) 相比,其选择性为67倍;与GMCSF (JAK2) 和EPO (JAK2) 信号传导 (IC50 >10,000 nM) 相比,选择性超过140倍。[2] 在人PBMC流式细胞术实验中,JAK3-IN-6 抑制IL-7诱导的CD3+, CD4+ T细胞中pSTAT5的IC50为280 nM。其选择性超过35倍,因为它不能强效抑制IL-6诱导的pSTAT3 (IC50 >9967 nM),并且对CD14+单核细胞中GMCSF诱导的pSTAT5抑制较弱 (IC50 = 9600 nM)。[2] 在一项评估4种细胞类型中9种细胞因子抑制情况的多重人全血流式细胞术实验中,JAK3-IN-6(在其对IL-7/pSTAT5的IC50浓度下)选择性地抑制了T细胞中IL-2、IL-7和IL-15(使用JAK1/JAK3的常见γ链细胞因子)的信号传导,但不抑制单核细胞或其他细胞类型中GMCSF、IL-3、IL-6、IL-10、IFNα或IFNγ的信号传导。[2] JAK3-IN-6 抑制大鼠全血中IL-7诱导的pSTAT5的IC50为1300 nM,但不抑制IL-6诱导的pSTAT3或GMCSF诱导的pSTAT5 (IC50 >20,000 nM)。[2] 在人PBMCs中,JAK3-IN-6 与一种JAK1选择性抑制剂在IL-7刺激下的交叉滴定实验表明,单独抑制JAK1或JAK3都足以完全抑制STAT5磷酸化,且它们的效应是相加的,而非协同的。[2] 在使用人CD34+来源的前红细胞进行的EPO依赖性增殖实验中,JAK3-IN-6 不抑制增殖 (IC50 >10,000 nM),证实了其对JAK2信号传导的选择性。[2] 在人CD4+ T细胞中进行的放线菌酮追踪实验测定,在正常血清条件下JAK3蛋白的半衰期 (τ½) 约为14.8小时,在血清饥饿条件下更快 (9.0小时)。1 μM JAK3-IN-6 的存在不改变JAK3蛋白的半衰期。[2] |
| 体内研究 (In Vivo) |
在预防性大鼠胶原诱导性关节炎模型中,口服给予JAK3-IN-6 (100、300、600 mg/kg,从第2天起每日一次) 可剂量依赖性地抑制足爪肿胀(AUC分别减少58%、92%和96%),并在300和600 mg/kg剂量下完全预防了踝关节骨矿物质密度损失。疗效与血液中IL-7/pSTAT5信号传导的时间加权平均抑制率相关(TWA抑制率分别为51%、70%和80%)。[2]
在治疗性大鼠CIA模型中(在第17天炎症建立后开始给药),JAK3-IN-6 与细胞色素P450抑制剂(ABT)联用以增加暴露量,产生了部分疗效。在300 mg/kg每日两次(达到高血浆暴露)时,其对足爪肿胀的抑制率为31%,并对骨质流失产生轻微但显著的抑制。[2] 在一项为期10天的大鼠研究中,JAK3-IN-6 (与ABT联用,100、300、600 mg/kg每日两次) 显著降低了总白细胞和淋巴细胞计数,但不影响单核细胞或中性粒细胞计数。即使在最高剂量下,它也不降低网织红细胞计数、红细胞计数、血红蛋白或血细胞比容,证实了其对JAK3(影响淋巴细胞)的选择性抑制优于JAK2(不影响红细胞生成)。[2] 一项大鼠离体PK/PD研究表明,单次口服300 mg/kg JAK3-IN-6 后,即使在给药后48小时血浆浓度较低(11 nM)时,血液中IL-7诱导的pSTAT5的抑制仍持续存在(抑制率17%),表明存在适度的延长药效学效应。离体IC50 (0.17 μM) 比相应的体外IC50低7.6倍。[2] |
| 酶活实验 |
使用HTRF(均相时间分辨荧光)法测定化合物对JAK亚型的效力(IC50)。将测试化合物在DMSO中连续稀释并加入检测板。加入含有特定JAK酶(例如155 pM JAK3)和生物素化肽底物的溶液,随后进行预孵育。通过加入接近酶Km浓度的ATP(例如JAK3为29.0 μM)来启动激酶反应。孵育120分钟后,使用含有EDTA、铕标记的抗磷酸酪氨酸抗体和链霉亲和素-dylight的检测缓冲液淬灭反应。进一步孵育后,使用TR-FRET检测(激发光320 nm,发射光比率665/615 nm)在酶标仪上读数。相对于未抑制对照计算抑制百分比,并通过曲线拟合确定IC50值。[2]
使用时间依赖性HTRF法测定JAK3-IN-6 对JAK3不可逆抑制的动力学参数(kinact, KI)。将化合物与JAK3酶预孵育不同时间。然后将预孵育混合物转移到含有ATP和肽底物的孔中启动反应。经过短暂反应期后,淬灭样品并按上述方法读数。根据剩余分数活性与预孵育时间关系的斜率确定每个抑制剂浓度下的酶失活观察速率常数(kobs)。通过绘制kobs与抑制剂浓度的关系图得到kinact和KI。[2] 使用跳释稀释HTRF法评估抑制的可逆性。将JAK3-IN-6 在高浓度下与JAK3预孵育,然后将混合物稀释100倍至含有ATP和底物的反应混合液中。监测酶活性随时间的变化。稀释后超过3小时未观察到JAK3活性恢复,证实了不可逆结合。[2] |
| 细胞实验 |
使用PathHunter β-半乳糖苷酶酶片段互补法评估单一JAK亚型的细胞活性。将表达全长JAK1、JAK2或JAK2/3嵌合体(包含JAK3的功能域,包括ATP结合口袋)并与β-gal的互补片段融合的工程化U2OS细胞铺板。加入化合物,然后用催乳素刺激以激活通路。通路激活后形成功能性β-gal,加入检测试剂后通过化学发光法测量其活性。化合物对信号传导的抑制以IC50量化。[2]
使用Cellsensor报告基因实验测量细胞因子通路抑制。使用稳定表达由特定STAT响应元件控制的β-内酰胺酶报告基因的细胞(例如,用于IL-2/STAT5的CTLL-2,用于GMCSF或EPO/STAT5的TF-1,用于IL-6/STAT3的ME180)。用化合物处理细胞,用相应的细胞因子(例如IL-2、GMCSF、IL-6)刺激,测量报告基因活性以确定化合物对通路抑制的IC50。[2] 通过磷酸化流式细胞术测量原代人细胞中STAT磷酸化的抑制情况。用人PBMCs或全血处理连续稀释的化合物,然后用特定细胞因子(例如IL-7、IL-6、GMCSF)刺激。固定、透化细胞,并用针对细胞表面标记物(CD3、CD4、CD14等)和磷酸化STAT(pSTAT5、pSTAT3)的荧光标记抗体染色。通过流式细胞术分析细胞,并使用特定细胞亚群(例如CD3+CD4+ T细胞)中pSTAT的中位荧光强度来生成剂量反应曲线并计算IC50值。[2] 对于EPO增殖实验,将人CD34+细胞分化为前红细胞。然后将这些细胞与连续稀释的化合物一起铺板,培养基中含有或不含EPO。4天后,使用基于荧光刃天青的染料(PrestoBlue)定量细胞活力/增殖。计算化合物对EPO依赖性增殖的影响。[2] IL-6诱导的MCP-1分泌实验使用人PBMCs。用化合物处理细胞,用IL-6刺激24小时,并使用基于电化学发光的免疫检测试剂盒测量上清液中分泌的MCP-1水平。[2] |
| 动物实验 |
对于预防性大鼠胶原诱导性关节炎 (CIA) 模型,雌性 Lewis 大鼠(7-8 周龄)于第 1 天在尾根部注射牛 II 型胶原蛋白(溶于弗氏不完全佐剂),并在第 8 天进行加强免疫。从第 2 天开始,大鼠每日口服 JAK3-IN-6(溶于 10% Tween 80)或载体,直至第 30 天。定期测量爪厚度。在第 29-30 天采集血液进行药代动力学 (PK) 分析。在实验结束时(第 30 天),处死动物,收集后肢进行骨矿物质密度微型 CT 分析,并采集血液进行临床化学/血液学检查。[2]
对于治疗性大鼠 CIA 模型,关节炎的诱导方法与上述相同。在爪肿胀出现后(第17天),开始给予JAK3-IN-6。为了增加化合物暴露量,将JAK3-IN-6(悬浮于10% Tween 80溶液中)与非特异性细胞色素P450抑制剂1-氨基苯并三唑(ABT,10 mg/kg,口服)共同口服给药。给药持续每日两次或按规定进行,直至第30天。爪肿胀测量、药代动力学取样和终末分析均采用类似方法进行。[2] 在为期10天的大鼠造血研究中,雌性Lewis大鼠连续给药10天。在第1天下午,口服给予ABT(或赋形剂)。从第2天到第11天,每天早上给予ABT(10 mg/kg,每日一次),随后立即口服JAK3-IN-6(100、300或600 mg/kg)或赋形剂。下午再给予一次化合物或赋形剂。在第10天和第11天,在不同时间点采集血液样本,用于药代动力学、血液学(全血细胞计数)和临床化学分析。[2] 在离体药代动力学/药效学研究中,Lewis大鼠单次口服给予JAK3-IN-6(300 mg/kg)。分别在给药后6、15、24和48小时处死各组大鼠。采集血液样本用于血浆药代动力学分析和离体流式细胞术检测。在离体实验中,用IL-7、IL-6或GM-CSF刺激全血,并测量特定细胞亚群中pSTAT的水平,以评估相对于血浆药物浓度的通路抑制情况。[2] |
| 药代性质 (ADME/PK) |
在预防性大鼠CIA研究中,每日一次口服100、300和600 mg/kg的JAK3-IN-6,其AUC(0-24h)值分别为38.5、92.1和164.4 μM·h。估计的Cmax值分别为4.8、12.3和11.7 μM。Tmax约为2小时。24小时谷浓度分别为0.11、0.26和2.04 μM。[2]在治疗性大鼠CIA研究中,JAK3-IN-6与ABT联合给药,该化合物的暴露量增加。在剂量分别为 10、30、100 和 300 mg/kg(与 ABT 联合给药)时,AUC(0-24h) 值分别为 11.1、23.0、80.1 和 199.6 μM·h。估计的 Cmax 值分别为 1.1、2.3、6.2 和 13.1 μM。Tmax 延迟至约 10-12 小时。[2]
在一项为期 10 天的大鼠造血研究中,JAK3-IN-6 与 ABT 联合给药,每日两次给药,剂量分别为 100、300 和 600 mg/kg,每日两次给药,其 AUC(0-24h) 值分别为 163.0、216.2 和 263.1 μM·h。 Cmax 值分别为 10.9、13.6 和 18.1 μM。Tmax 为 11-15 小时。Ctrough 值分别为 1.1、2.6 和 2.05 μM。[2] 据报道,JAK3-IN-6 在大鼠体内的血浆半衰期 (t1/2) 约为 0.3 小时。[2] |
| 毒性/毒理 (Toxicokinetics/TK) |
在为期10天的大鼠研究中,JAK3-IN-6(与ABT联用,剂量高达600 mg/kg,每日两次)显著降低了白细胞和淋巴细胞计数,这与其抑制JAK3依赖性细胞因子信号通路(例如IL-2、IL-7)的机制相符,而这些信号通路对于淋巴细胞的存活/增殖至关重要。这被认为是药理学介导的靶向效应,而非一般毒性。[2]
JAK3-IN-6不会引起贫血或影响红细胞生成。即使在最高剂量下,它也没有显著降低网织红细胞计数、红细胞计数、血红蛋白或血细胞比容,这表明其对JAK2具有选择性,并且避免了与泛JAK抑制相关的造血系统不良事件。[2] |
| 参考文献 | |
| 其他信息 |
JAK3-IN-6(化合物 2) 是一种不可逆抑制剂,其设计目的是通过丙烯酰胺弹头与 JAK3 激酶结构域中的 Cys-909 共价结合,从而减轻因 JAK 同工酶之间 ATP Km 差异而导致的细胞效力变化,而这种变化困扰着可逆抑制剂。[2]
该化合物被开发为评估自身免疫性疾病中 JAK3 生物学的工具。研究表明,单独选择性地药理抑制JAK3足以抑制通过共同γ链细胞因子受体(例如IL-2、IL-7、IL-15)的信号传导,这些受体利用JAK1/JAK3异二聚体。[2] 该研究提示,虽然像JAK3-IN-6这样的选择性JAK3抑制剂在预防性治疗中(例如在大鼠胶原诱导性关节炎模型中)可以完全有效地阻断炎症的发展,但在逆转已形成的炎症(治疗性治疗)中可能只有部分疗效,因为疾病后期阶段涉及JAK3非依赖性细胞因子。[2] 数据支持以下假设:选择性JAK3抑制剂可以在类风湿性关节炎等自身免疫性疾病中保持疗效,同时可能避免与JAK2抑制(例如贫血)和JAK1抑制(例如血脂异常、感染风险增加)相关的不良事件。[2] JAK3的相对快速周转人类T细胞中的JAK3蛋白(半衰期约为14.8小时,在激活/应激条件下半衰期更快)可能会限制不可逆JAK3抑制剂在临床炎症环境下所能达到的长期药效作用。[2] |
| 分子式 |
C19H18N4O3
|
|
|---|---|---|
| 分子量 |
350.371223926544
|
|
| 精确质量 |
350.137
|
|
| CAS号 |
1443235-95-7
|
|
| 相关CAS号 |
|
|
| PubChem CID |
89629876
|
|
| 外观&性状 |
White to light yellow solid powder
|
|
| LogP |
2.7
|
|
| tPSA |
97
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
5
|
|
| 可旋转键数目(RBC) |
6
|
|
| 重原子数目 |
26
|
|
| 分子复杂度/Complexity |
553
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
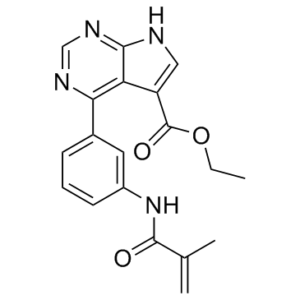
O(CC)C(C1=CNC2C1=C(C1C=CC=C(C=1)NC(C(=C)C)=O)N=CN=2)=O
|
|
| InChi Key |
NJPHKVOXHRAPGJ-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C19H18N4O3/c1-4-26-19(25)14-9-20-17-15(14)16(21-10-22-17)12-6-5-7-13(8-12)23-18(24)11(2)3/h5-10H,2,4H2,1,3H3,(H,23,24)(H,20,21,22)
|
|
| 化学名 |
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.17 mg/mL (6.19 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 21.7 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.17 mg/mL (6.19 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 21.7 mg/mL澄清DMSO储备液加入900 μL玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8541 mL | 14.2706 mL | 28.5413 mL | |
| 5 mM | 0.5708 mL | 2.8541 mL | 5.7083 mL | |
| 10 mM | 0.2854 mL | 1.4271 mL | 2.8541 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Predicted cellular potency of compound 1 confirmed by potency in single isozyme cellular assay.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
Potent, selective irreversible JAK3 inhibitor 2. (A) Compound 2 structure. (B) Confirmation of covalent binding by X-ray crystallography. (C) Waterfall plot demonstrating selectivity of compound 2 against the kinome. |
Fractional activity remaining and relative affinity (kinact/KI) determination.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
Multiparameter flow cytometry in human whole blood demonstrates pSTAT signature of compound 2 specific to JAK3 signaling.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
No synergy observed in pSTAT5 PBMC assay in response to IL-7 with cross-titrated JAK1 and JAK3 inhibitors.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
Prophylactic inhibition of JAK3 in a rat CIA model blocks paw swelling and joint degradation.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
Therapeutic inhibition of JAK3 in a rat CIA model partially blocks paw swelling and joint degradation.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
Inhibition of leukocyte proliferation, but not erythrocyte proliferation, by compound 2 in rats.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
Ex vivo pSTAT inhibition by compound 2 in rat whole blood. pSTAT signal in whole blood obtained from rats dosed with 300 mg/kg of compound 2 and stimulated with cytokines ex vivo.J Pharmacol Exp Ther.2017 May;361(2):229-244. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA









 463611831
463611831
