| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| Other Sizes |
|

描述: 洛诺昔康(氯替诺昔康;Xefo;Lornoxicamum;Xefocam;TS-110;Ro 13-9297;TS110)是一种非甾体抗炎药 (NSAID),是强效的 COX-1/COX-2 抑制剂,具有潜在的抗炎活性。它已被批准用于治疗疼痛、骨关节炎和类风湿性关节炎。洛诺昔康是一种新型的非甾体抗炎药 (NSAID),属于昔康类药物。通过强效抑制COX-1和COX-2,该药物(口服和注射剂型)均表现出显著的镇痛、抗炎和解热作用。
| 靶点 |
Cyclooxygenase (COX) (inhibits prostaglandin synthesis via inhibition of COX, but does not inhibit 5-lipoxygenase)
COX-1 and COX-2 (selectivity ratio of human COX-1:COX-2 inhibitory potency = 0.6, intermediate between tenidap or piroxicam (0.2) and tenoxicam (1.2) or ketorolac (1.3)) [1] |
|---|---|
| 体外研究 (In Vitro) |
体外活性:洛诺昔康在缓解妇科或骨科手术后的疼痛方面,与阿片类镇痛药吗啡、哌替啶(美哌利啶)和曲马多一样有效;在口腔手术后的疼痛缓解方面,其疗效与其他非甾体抗炎药(NSAIDs)相当。洛诺昔康在缓解骨关节炎、类风湿性关节炎、强直性脊柱炎、急性坐骨神经痛和腰痛的症状方面,也与其他非甾体抗炎药一样有效。 |
| 体内研究 (In Vivo) |
洛诺昔康剂量依赖性地减少c-Fos-LI神经元的总数,最高剂量9 mg/kg时减少最为显著,达到75%,而低剂量0.3 mg/kg时减少45%。洛诺昔康(0.1、0.3 mg/kg、1 mg/kg、3 mg/kg和9 mg/kg,静脉注射)显著减少了脊髓背角浅层(分别减少24%、33%、53%、54%和63%)和深层(分别减少28%、48%、62%、69%和79%)的c-Fos-LI神经元数量。在慢性关节炎大鼠模型中,洛诺昔康的有效剂量(ED50)分别为0.083 mg/kg、3.9 mg/kg和4.3 mg/kg,可有效降低痛觉过敏。洛诺昔康可显著降低大鼠爪渗出液和脑脊液中的PGE2水平。在急性水肿大鼠中,洛诺昔康0.16 mg/kg、塞来昔布4 mg/kg和洛索洛芬2.4 mg/kg均能显著降低痛觉过敏,且效果相似。体内实验:在小鼠乙酰胆碱诱导扭体试验中,洛诺昔康的镇痛活性约为替诺昔康的10倍(洛诺昔康口服ED50 = 0.1 mg/kg)。 [1]体内实验:在角叉菜胶诱导的大鼠水肿试验中,洛诺昔康的活性是替诺昔康的10倍(洛诺昔康口服ED50为0.4 mg/kg,替诺昔康为4 mg/kg)。[1]体内实验:在佐剂诱导的多关节炎大鼠模型中,洛诺昔康抑制爪肿胀的活性是替诺昔康的10倍(洛诺昔康口服ED50为0.03 mg/kg,替诺昔康为0.3 mg/kg)。口服0.1 mg/kg的洛诺昔康可预防退行性骨丢失,其机制可能是抑制多形核白细胞迁移。 [1]体内实验:洛诺昔康在达到抗炎作用所需剂量的10倍时,仍表现出解热作用。[1]体内实验:在花生四烯酸诱导的小鼠致死模型中,洛诺昔康的效力高于吲哚美辛和吡罗昔康(洛诺昔康口服ED50为0.012 mg/kg,吲哚美辛为0.150 mg/kg,吡罗昔康为0.870 mg/kg)。[1]
|
| 酶活实验 |
酶活性测定:通过体外测定大鼠多形核白细胞中PGD2的生成量,评估了洛诺昔康对前列腺素合成的抑制作用。测定了50%抑制浓度(IC50)。洛诺昔康的IC50为0.02 μmol/L,而替诺昔康的IC50为2.45 μmol/L。[1] 酶活性测定:基于人COX-1与COX-2抑制效力的比值,确定了洛诺昔康对COX-1与COX-2的选择性,该比值为0.6(数据存档)。[1]
|
| 细胞实验 |
细胞实验:采用大鼠多形核白细胞测定PGD2生成量,以此作为环氧合酶活性的指标。将细胞与洛诺昔康孵育,并计算抑制PGD2生成的IC50值。[1]
细胞实验:采用人多形核白细胞研究超氧化物释放;洛诺昔康抑制超氧化物释放。采用人血小板研究血小板衍生生长因子(PDGF)释放;洛诺昔康抑制PDGF释放。[1] 细胞实验:采用猪软骨培养物评估蛋白聚糖合成。浓度为0.1至1.0 mg/L的洛诺昔康刺激蛋白聚糖合成。[1] |
| 动物实验 |
0.1、0.3 mg/kg、1 mg/kg、3 mg/kg 和 9 mg/kg,静脉注射大鼠
动物实验方案:乙酰胆碱诱导小鼠扭体试验:口服不同剂量的洛诺昔康,以确定抑制扭体反应的 ED50 值。[1] 动物实验方案:角叉菜胶诱导大鼠水肿试验:口服洛诺昔康,测量爪水肿,计算抑制作用的 ED50 值。[1] 动物实验方案:佐剂诱导大鼠多发性关节炎:口服洛诺昔康,评估爪肿胀和骨丢失情况。确定抑制爪肿胀的 ED50 值。 [1] 动物实验方案:花生四烯酸诱导小鼠致死性:口服洛诺昔康,并计算保护50%动物的有效剂量(ED50)。[1] 动物实验方案:健康志愿者内镜研究:采用交叉设计,每日给予洛诺昔康16 mg,连续7天,并与萘普生1000 mg/天进行比较,评估胃十二指肠损伤。[1] 动物实验方案:健康志愿者粪便失血研究:每日两次给予洛诺昔康4 mg,连续14天,并与吲哚美辛50 mg,每日两次进行比较。[1] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
洛诺昔康经胃肠道迅速且几乎完全吸收(90-100%)。代谢/代谢物:洛诺昔康完全由CYP2C9代谢,主要代谢物为5'-羟基洛诺昔康。仅有极少量的完整洛诺昔康以原形经尿液排出。约三分之二的药物以活性形式经肝脏排泄,三分之一经肾脏排泄。洛诺昔康的已知代谢物包括5'-羟基洛诺昔康。生物半衰期:3-5小时 ADME/药代动力学:吸收:洛诺昔康口服后完全吸收,4 mg剂量后2.5小时内(tmax)血浆峰浓度(Cmax)达到270 μg/L。食物会延迟吸收并略微降低(约20%)。肌注后的生物利用度估计为87%。[1] 分布:洛诺昔康与血浆蛋白(几乎全部为血清白蛋白)高度结合(99%),表观分布容积较小(0.2 L/kg)。它能渗透到血管周围组织间隙,包括滑液。类风湿性关节炎患者每日两次,每次4 mg,连续服用5天后,滑液与血浆的AUC比值约为0.5。[1] 代谢:洛诺昔康主要在肝脏中代谢,主要通过细胞色素P450 (CYP)2C亚家族(可能还有CYP2C9)代谢为无活性代谢物5'-羟基洛诺昔康。尿液中检测到的原药量极少。 [1] 排泄:单次口服4 mg [14C]-洛诺昔康后,42%的放射性标记物经尿液排出,51%经粪便排出。原形药物的肾清除率可忽略不计。[1] 半衰期:在健康年轻志愿者中,洛诺昔康的末端血浆消除半衰期 (t½β) 相对较短,为3至5小时,但个体间差异较大。5'-羟基代谢物的半衰期约为11小时。[1] 清除率:在健康志愿者中,洛诺昔康(每日两次,每次4至8 mg)的表观口服清除率范围为1.5至3.4 L/h。 [1] 年龄/疾病的影响:药物动力学不会因高龄或肾功能损害而发生显著改变,但肝功能受损的患者体内会出现非活性代谢物的蓄积。在严重肾功能不全的情况下,肠肝排泄增强可能补偿肾脏排泄减少。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
蛋白结合
洛诺昔康与血浆蛋白的结合率为99%(几乎完全与血清白蛋白结合)。 毒性/毒代动力学:胃肠道效应:在健康志愿者中,每日服用16 mg洛诺昔康引起的内镜证实的胃十二指肠损伤显著低于每日服用1000 mg萘普生。每日服用8 mg洛诺昔康引起的粪便出血量往往低于每日服用100 mg吲哚美辛。在18例患者中,有17例每日两次服用4 mg洛诺昔康,持续2周,未发现血清胃蛋白酶原I水平升高。[1] 肾脏效应:短期研究(≤3周)显示,每日两次服用高达8 mg的洛诺昔康,在健康志愿者或轻度至中度肾功能损害患者中未发现肾毒性。 [1] 血液学效应:健康志愿者每日两次服用4 mg洛诺昔康,持续14天,除延长血栓形成起始时间(从489秒延长至567秒,p<0.01)外,未对止血、凝血或溶栓产生影响。[1] 蛋白结合:洛诺昔康与血浆蛋白的结合率为99%。[1] 药物相互作用:西咪替丁抑制洛诺昔康的消除(使Cmax增加28%,AUC增加9%,血浆清除率降低12%)。雷尼替丁和抗酸剂(铝、镁、钙、铋)对其药代动力学无明显影响。洛诺昔康可使华法林血浆浓度升高约30%,并增强其抗凝作用。它可使稳态血清锂浓度峰值(Cmax)增加20%。它可减弱呋塞米的利尿和利钠作用。它可使甲氨蝶呤的药时曲线下面积(AUC)增加20%以上。它可增强格列本脲的代谢作用(24小时血浆胰岛素分泌增加约45%),但不影响其药代动力学。对非那宗(安替比林)的清除率无影响,表明其对肝脏药物代谢酶无影响。[1] |
| 参考文献 |
Drugs.1996 Apr;51(4):639-57;Inflammopharmacology.1997;5(4):331-41.
|
| 其他信息 |
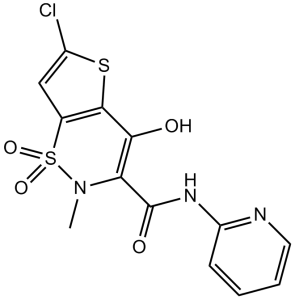
洛诺昔康是一种噻吩昔康单羧酸酰胺,由6-氯-4-羟基-2-甲基噻吩并[2,3-e][1,2]噻嗪-3-羧酸1,1-二氧化物的羧基与2-氨基吡啶的氨基缩合而成。它主要用于治疗关节炎症、骨关节炎、术后疼痛、坐骨神经痛和其他炎症性疼痛。它是一种非甾体类抗炎药(NSAID),具有非麻醉性镇痛和解热作用。它属于噻吩昔康类、吡啶类、单羧酸酰胺类、有机氯化合物和杂芳基羟基化合物。洛诺沙(氯诺沙)是一种新型的氧沙星类非甾体抗炎药,具有镇痛、抗炎和解热作用。洛诺沙星与其他氧沙星类化合物的不同之处在于其对前列腺素生物合成的强效抑制作用,这一特性解释了其显著的治疗效果。洛诺沙星已在日本获准上市。
洛诺沙星是一种高生物利用度的口服非甾体抗炎药(NSAID),属于环氧合酶类,具有镇痛、解热、抗血栓和抗炎作用。口服后,洛诺沙星与环氧合酶(COX)1型(COX-1)和2型(COX-2)结合并抑制其活性。该药阻断COX介导的信号通路,从而减少前列腺素和血栓素的生成,缓解疼痛、发热和炎症。 适应症 用于治疗急性轻度至中度疼痛,以及某些类型风湿性疾病引起的关节炎症和疼痛。 作用机制 与其他非甾体抗炎药 (NSAIDs) 一样,洛诺昔康的抗炎和镇痛作用与其通过抑制 COX-1 和 COX-2 来抑制前列腺素和血栓素的合成有关。这可以减轻前列腺素介导的炎症、疼痛、发热和肿胀。然而,与其他非甾体抗炎药 (NSAIDs) 一样,洛诺昔康的确切作用机制尚未完全阐明。 药效学 洛诺昔康是一种属于氧沙星类的非甾体抗炎药 (NSAID)。与其他 NSAIDs 一样,洛诺昔康是环氧合酶的强效抑制剂,环氧合酶催化花生四烯酸转化为前列腺素(在炎症过程中作为信使分子)和血栓素。与某些非甾体抗炎药不同,洛诺昔康对环氧合酶的抑制作用不会导致白三烯生成增加,这意味着花生四烯酸不会进入5-脂氧合酶级联反应,从而最大限度地降低不良反应的风险。 附加信息:作用机制:除了抑制环氧合酶外,洛诺昔康可能增加内源性阿片类物质的释放;在急性腰痛患者中,静脉注射洛诺昔康4毫克,每日两次,持续5天,可显著提高血浆中强啡肽(增加140%)和β-内啡肽(增加33%)的水平。 [1] 治疗潜力:洛诺昔康已被评估用于治疗急性疼痛(骨科/妇科手术后、口腔手术后、急性坐骨神经痛/腰痛)和慢性疼痛(骨关节炎、类风湿性关节炎、强直性脊柱炎、慢性腰痛、偏头痛预防)。其在术后镇痛方面与哌替啶、曲马多和吗啡疗效相当,在治疗关节炎方面与双氯芬酸、萘普生、吡罗昔康和吲哚美辛疗效相当。[1] 剂量:常用口服剂量为:关节炎和腰痛患者每日两次或三次,每次4毫克;或每日两次,每次8毫克;术后镇痛患者可单次或多次服用4毫克或8毫克(口服或静脉注射)。 [1] 注意:不建议肾功能受损的患者以及正在服用华法林、口服磺脲类药物、袢利尿剂或噻嗪类利尿剂或地高辛的患者使用。[1] |
| 分子式 |
C13H10CLN3O4S2
|
|
|---|---|---|
| 分子量 |
371.82
|
|
| 精确质量 |
370.98
|
|
| CAS号 |
70374-39-9
|
|
| 相关CAS号 |
Lornoxicam-d4;1216527-48-8
|
|
| PubChem CID |
54690031
|
|
| 外观&性状 |
Light yellow to yellow solid powder
|
|
| 密度 |
1.7±0.1 g/cm3
|
|
| 熔点 |
225-230°C (dec.)
|
|
| 折射率 |
1.741
|
|
| LogP |
2.18
|
|
| tPSA |
136.22
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
23
|
|
| 分子复杂度/Complexity |
634
|
|
| 定义原子立体中心数目 |
0
|
|
| InChi Key |
WLHQHAUOOXYABV-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C13H10ClN3O4S2/c1-17-10(13(19)16-9-4-2-3-5-15-9)11(18)12-7(23(17,20)21)6-8(14)22-12/h2-6,18H,1H3,(H,15,16,19)
|
|
| 化学名 |
6-chloro-4-hydroxy-2-methyl-1,1-dioxo-N-pyridin-2-ylthieno[2,3-e]thiazine-3-carboxamide
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6895 mL | 13.4474 mL | 26.8947 mL | |
| 5 mM | 0.5379 mL | 2.6895 mL | 5.3789 mL | |
| 10 mM | 0.2689 mL | 1.3447 mL | 2.6895 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05679453 | Completed | Drug: Lornoxicam 8 Mg Oral Tablet | Pain, Acute Edema Trismus |
Kutahya Health Sciences University | July 20, 2022 | Phase 4 |
| NCT00997750 | Completed | Drug: Lornoxicam | Acute Coronary Syndrome | Central Clinical Hospital w/Outpatient Health Center of Business Administration for the President of Russian Federation | March 2007 | Phase 4 |
| NCT02750917 | Completed | Drug: Lornoxicam Drug: Etoricoxib |
Postoperative Pain | Foisor Orthopedics Clinical Hospital | September 2014 | Phase 3 |
| NCT01117948 | Terminated Has Results | Drug: Lornoxicam Drug: Placebo |
Alzheimer´s Disease | JSW Lifesciences | September 2009 | Phase 2 |
| NCT01055470 | Completed | Drug: Diclofenac Drug: Lornoxicam |
Osteoarthritis of Knee Joint | Government Medical College, Bhavnagar | December 2008 | Not Applicable |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
