| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
PKC-α (IC50 = 5 nM); PKC-βII (IC50 = 14 nM); MAPKAP-K1b (IC50 = 3 nM); MSK1 (IC50 = 8 nM); S6K1 (IC50 = 15 nM);PKC-βI (IC50 = 24 nM); PKC-ε (IC50 = 24 nM); PKC-γ (IC50 = 27 nM); Rat Brain PKC (IC50 = 23 nM); GSK3β (IC50 = 38 nM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
Ro 31-8220 是一种强效 PKC 抑制剂,对 PKCα、PKCβI、PKCβII、PKCγ、PKCε 和大鼠脑 PKC 的 IC50 值分别为 5、24、14、27、24 和 23 nM[1]。此外,Ro 31-8220 强烈抑制 MSK1、S6K1、GSK3β 和 MAPKAP-K1b(IC50 分别为 3、8、15 和 38 nM); MKK3、MKK4、MKK6 和 MKK7 不受影响。此外,电压依赖性 Na+ 通道直接被 Ro 31-8220[2] 抑制。在小脑颗粒神经元中,Ro 31-8220 (1 μM) 对对氧磷诱导的神经元细胞死亡具有神经保护作用,抑制对氧磷诱导的 caspase-3 活性,并降低对氧磷引起的磷酸 PKC pan 水平的增加 [3]。
|
||
| 体内研究 (In Vivo) |
在小鼠中,Ro 31-8220(6 mg/kg/d,皮下注射)的半衰期为 5.7 小时,且耐受性良好。经过六周的治疗后,用 Ro 31-8220 治疗的 MLP−/− 小鼠在缩短分数方面表现出显着的改善,但 WT 小鼠没有表现出任何变化[4]。
药理抑制PKCα可恢复MLP−/−小鼠的心功能[4] 由于传统的肌力药物慢性治疗与心力衰竭患者的不良结果相关,因此我们在这里研究了慢性给予Ro-31-8220超过4-6周对MLP - / -心力衰竭小鼠的影响。在研究开始时和6周后,通过超声心动图评估所有小鼠的心室功能。Ro-31-8220(或对照品)每天注射一次,剂量为6mg /kg/day, s.q。Ro-31-8220化合物被用于Ro-32-0432化合物的所有体内研究,只是因为与大规模合成相关的费用问题以及在小鼠中长期使用所需的量。Ro-31-8220在大鼠体内的药动学分析 药物的半衰期为5.7小时,6小时时血浆浓度比PKCα的IC50高100倍(见讨论)。该剂量的Ro-31-8220在小鼠中耐受性良好,在6周内没有观察到的有害影响,也不影响体重。在6周的时间内,注射了载体或Ro-31-8220的野生型小鼠在分数缩短或任何其他心室尺寸测量方面没有变化(图5A和表1)。相比之下,与载体治疗或治疗前的基线值相比,6周注射Ro-31-8220的MLP - / -小鼠在分数缩短方面表现出显著的恢复(图5A,表1)。图5A所示的研究是在6个月大的野生型和MLP - / -小鼠中进行的。尽管在4周的治疗中,老龄MLP−/−小鼠(14个月)与给药小鼠相比,在部分缩短方面也有类似的改善(图5B,表1)。野生型和MLP−/−小鼠也接受了分离的、工作的心脏准备,显示长期给药Ro-31-8220的MLP−/−小鼠的心脏收缩力显著增加。与野生型小鼠相当(表2)。在舒张功能(- dP/dt)和左心室压力方面也观察到类似的恢复(表2)。 虽然Ro-31-8220在4周或6周的慢性给药期间挽救了MLP - / -小鼠的心脏收缩性能,但它没有改变心脏重量,也没有通过超声心动图评估逆转心室扩张,也没有改善组织病理学(数据未显示)。这些观察结果表明,与慢性Ro-31-8220治疗相关的心脏功能的部分增加可能涉及对收缩力本身的急性影响。事实上,在MLP−/−小鼠中注射Ro-31-8220仅3天,与载药治疗相比,分数缩短得到改善(图5C)。 |
||
| 酶活实验 |
蛋白激酶C(PKC)同工酶家族被认为在许多不同的细胞类型中介导了广泛的信号转导途径。一系列双吲哚基马来酰亚胺已被评估为传统PKC家族(PKCsα、β、γ)成员的抑制剂,以及新的Ca(2+)非依赖性PKC家族PKCε的代表性抑制剂。与吲哚咔唑星孢菌素相比,所有研究的双吲哚基马来酰亚胺对PKCα的选择性都比其他检测的同工酶低。此外,带有构象限制侧链的双吲哚基马来酰亚胺作为PKCε抑制剂的活性较低。其中最引人注目的是Ro 32-0432,它对PKCα的选择性是PKCε的10倍,对PKCβI的选择性是PKC-ε的4倍[2]。
|
||
| 细胞实验 |
对硫磷的活性代谢产物对氧磷是一种乙酰胆碱酯酶(AChE)抑制剂,通过凋亡机制杀死培养的小脑颗粒细胞神经元。蛋白激酶C是一种具有多种功能的酶,但其在对氧磷诱导的细胞死亡中的作用尚不清楚。我们发现,神经毒性浓度的对氧磷会增加PKC磷酸化。我们使用PKC激活剂佛波醇12-肉豆蔻酸13-乙酸酯(TPA)测试了PKC是否参与对氧磷诱导的神经元细胞死亡。TPA可增加PKC活性,并将对氧磷的神经毒性作用增强28%。与此形成鲜明对比的是,添加PKC抑制剂Ro-31-8220可以保护30%以上的神经元,否则这些神经元在治疗前或治疗后都会死于对氧磷诱导的神经元细胞死亡,并显著降低磷酸化PKC水平。研究还表明,Ro-31-8220的预处理完全阻断了对氧磷诱导的半胱氨酸天冬氨酸蛋白酶-3活性。这些结果表明,对氧磷神经毒性需要激活蛋白激酶C。[3]
在体外(DIV)8的指定时间内,将神经毒性浓度的对氧磷(200μM)添加到颗粒细胞培养物中。在DIV 8暴露于对氧磷之前或之后,将以下药物添加到颗粒细胞培养物中:在添加对氧磷前15分钟或后3小时添加Ro-81-3220(1μM)。在加入对氧磷前15分钟加入TPA(0.1μM)[1]。 |
||
| 动物实验 |
|
||
| 参考文献 |
|
||
| 其他信息 |
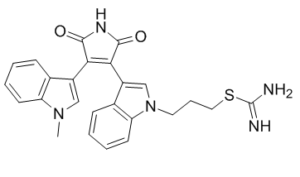
Ro 31-8220 是一种亚氨基硫代氨基甲酸酯,属于吲哚类和马来酰亚胺类化合物。它是一种 EC 2.7.11.13(蛋白激酶 C)抑制剂。其功能与马来酰亚胺类似。
Ro 31-8220 是一种甲磺酸盐形式,是星形孢菌素类似物,可抑制蛋白激酶 C 并诱导细胞凋亡。 蛋白激酶 C (PKC) 同工酶家族被认为介导多种不同细胞类型中的广泛信号转导通路。一系列双吲哚基马来酰亚胺已被评估为传统 PKC 家族成员(PKC-α、-β、-γ)以及新型 Ca2+ 非依赖性 PKC 家族代表 PKC-ε 的抑制剂。与吲哚咔唑类化合物星形孢菌素相比,所有研究的双吲哚基马来酰亚胺类化合物对 PKC-α 的选择性均略高于其他同工酶。此外,具有构象受限侧链的双吲哚基马来酰亚胺类化合物对 PKC-ε 的抑制活性较低。其中最显著的是 Ro 32-0432,其对 PKC-α 的选择性是 PKC-ε 的 10 倍,对 PKC-β I 的选择性是 PKC-ε 的 4 倍。[1] 我们检测了 28 种市售化合物的特异性,这些化合物据报道是特定丝氨酸/苏氨酸特异性蛋白激酶的相对选择性抑制剂,并考察了它们对多种蛋白激酶的抑制作用。化合物KT 5720、Rottlerin和槲皮素被发现能够抑制多种蛋白激酶,有时其抑制效力甚至远超其假定的靶标,因此基于这些化合物在细胞实验中得出的结论可能存在误差。Ro 318220及其相关的双吲哚马来酰亚胺类化合物,以及H89、HA1077和Y 27632,虽然是更具选择性的抑制剂,但仍能以相似的效力抑制两种或两种以上的蛋白激酶。LY 294002被发现能够以与磷脂酰肌醇3-激酶相似的效力抑制酪蛋白激酶-2。选择性最显著的化合物包括KN62、PD 98059、U0126、PD 184352、雷帕霉素、沃特曼宁、SB 203580和SB 202190。与PD 98059类似,U0126和PD 184352在细胞实验中通过抑制丝裂原活化蛋白激酶(MAPK)激酶(MKK1)的活化而非直接抑制MKK1活性来阻断MAPK级联反应。除雷帕霉素和PD 184352外,即使是选择性最高的抑制剂也至少会影响一种其他蛋白激酶。我们的结果表明,不能仅通过研究蛋白激酶抑制剂对一级结构密切相关的激酶的影响来评估其特异性。我们提出了在基于细胞的检测中使用蛋白激酶抑制剂的指导原则。[2] 对氧磷是硫磷的活性代谢产物,是一种乙酰胆碱酯酶 (AChE) 抑制剂,它通过凋亡机制杀死培养的小脑颗粒细胞。蛋白激酶 C (PKC) 是一种具有多种功能的酶,但其在对氧磷诱导的细胞死亡中的作用尚不清楚。我们发现,神经毒性浓度的对氧磷会增加 PKC 的磷酸化。我们使用 PKC 激活剂佛波醇 12-肉豆蔻酸酯 13-乙酸酯 (TPA) 来检测 PKC 是否参与对氧磷诱导的神经元细胞死亡。TPA 可增加 PKC 活性,并使对氧磷的神经毒性作用增强 28%。与此形成鲜明对比的是,添加 PKC 抑制剂 Ro-31-8220 可保护超过 30% 的神经元免于因对氧磷诱导的神经元细胞死亡而死亡,无论是在预处理还是后处理模式下,并且显著降低了磷酸化 PKC 的总体水平。我们还发现,Ro-31-8220 的预处理可完全阻断对氧磷诱导的 caspase-3 活性。这些结果表明,蛋白激酶 C 的激活是导致对氧磷神经毒性的必要条件。[3] 背景:传统的蛋白激酶 C (PKC) 亚型 α 在心肌细胞中作为 Ca2+ 处理的近端调节因子发挥作用。小鼠中 PKCα 的缺失会导致肌浆网 Ca2+ 负荷增加、Ca2+ 瞬变增强和收缩力增强,而心脏中 PKCα 的过表达则会减弱收缩力。从机制上讲,PKCα通过改变抑制剂-1的磷酸化状态直接调节Ca2+处理,进而抑制蛋白磷酸酶-1的活性,从而调节磷蛋白的活性,并继而调节肌浆网Ca2+ ATPase的活性。方法和结果:在本研究中,我们发现,在野生型小鼠体内或离体做功心脏模型中,使用Ro-32-0432或Ro-31-8220短期抑制常规PKC亚型可显著增强心脏收缩力,但在PKCα缺陷小鼠中未观察到此现象。Ro-32-0432在两种不同的体内心力衰竭模型中也增强了心脏收缩力。在肌肉lim蛋白基因缺失导致的心力衰竭小鼠模型中,短期或长期使用Ro-31-8220治疗可显著增强心脏收缩力并恢复泵血功能。此外,在心肌梗死后心肌病大鼠模型中,使用显性负性 PKCα cDNA 进行腺病毒介导的基因治疗可挽救心力衰竭。研究还发现,PKCα 是成人心脏中表达的主要经典 PKC 亚型,这表明这些发现可能与人类病理生理学相关。结论:药理学抑制 PKCα 或一般经典亚型可能成为增强某些阶段心力衰竭患者心肌收缩力的新型治疗策略。[4] |
| 分子式 |
C25H23N5O2S
|
|
|---|---|---|
| 分子量 |
457.55
|
|
| 精确质量 |
457.157
|
|
| CAS号 |
125314-64-9
|
|
| 相关CAS号 |
Ro 31-8220 mesylate;138489-18-6
|
|
| PubChem CID |
5083
|
|
| 外观&性状 |
Typically exists as
Pink to red solid at room temperature
|
|
| 密度 |
1.42g/cm3
|
|
| LogP |
5.43
|
|
| tPSA |
193.95
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
33
|
|
| 分子复杂度/Complexity |
845
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
S(/C(=N/[H])/N([H])[H])C([H])([H])C([H])([H])C([H])([H])N1C([H])=C(C2C(N([H])C(C=2C2=C([H])N(C([H])([H])[H])C3=C([H])C([H])=C([H])C([H])=C23)=O)=O)C2=C([H])C([H])=C([H])C([H])=C12
|
|
| InChi Key |
DSXXEELGXBCYNQ-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C25H23N5O2S/c1-29-13-17(15-7-2-4-9-19(15)29)21-22(24(32)28-23(21)31)18-14-30(11-6-12-33-25(26)27)20-10-5-3-8-16(18)20/h2-5,7-10,13-14H,6,11-12H2,1H3,(H3,26,27)(H,28,31,32)
|
|
| 化学名 |
3-[3-[4-(1-methylindol-3-yl)-2,5-dioxopyrrol-3-yl]indol-1-yl]propyl carbamimidothioate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1856 mL | 10.9278 mL | 21.8555 mL | |
| 5 mM | 0.4371 mL | 2.1856 mL | 4.3711 mL | |
| 10 mM | 0.2186 mL | 1.0928 mL | 2.1856 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA


 463611831
463611831
