| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
MOR ( EC50 = 97 nM )
μ-Opioid Receptor (μOR) (Ki = 14 nM; EC₅₀ for G protein activation = 31 nM; EC₅₀ for β-arrestin 2 recruitment = 144 nM; Bias Factor = 17 [G protein/β-arrestin 2]) [1] |
||
|---|---|---|---|
| 体外研究 (In Vitro) |
体外活性:SR17018 是一种 µ-阿片受体 (MOR) 激动剂,与 GTPγS 结合,EC50 为 97 nM。 SR17018 在低于 10 μM 时对诱导 βarrestin2 募集至 MOR 没有明显影响。 SR17018 通过 G 蛋白或 βarrestin2 促进信号传导。激酶测定:为了确定 βarrestin2 招募到人 MOR 的情况,使用了商业酶片段互补测定(β-半乳糖苷酶)。 16-20 小时前,将 U2OS-βarrestin-hMOR PathHunter 细胞以每孔 5,000 个细胞的密度接种在 384 孔白壁测定微孔板 (Greiner Bio-One) 中的 Assay Complete Cell Placing 5 Reagent (DiscoveRx) 中。测量信号。在 37°C 下,用浓度逐渐增加的测试化合物处理细胞 90 分钟,并使用 PathHunter 检测试剂盒和 β-半乳糖苷酶底物测定 βarrestin2 的募集情况,以检测功能性 β-半乳糖苷。使用 SpectraMax M5e 酶标仪 (Molecular Devices) 测量由此产生的发光增加。 PathHunter U2OS OPRM1 βarrestin 细胞的平均载体为 446 ± 25 RLU,DAMGO 的平均载体倍数为 36 ± 1。 细胞测定:CHO-hMOR、-hDOR 和 -hKOR 细胞以每 4,000 个细胞的密度进行铺板。测定前 4 小时,将 384 孔、白壁、30 μl 体积微孔板 (Greiner Bio-One) 的孔置于含有 1% FBS 的 Opti-MEM 中。将细胞用 20 μM 毛喉素、25 μM 4-(3-丁氧基-4-甲氧基苯甲基)咪唑烷-2-酮 (Ro-20-1724) 和浓度不断增加的测试化合物在 25°C 下处理 30 分钟。然后使用 Cisbio (Cisbio-62AM6PEC) 的均质时间分辨荧光共振能量转移 (FRET) cAMP HiRange 测定法测定 cAMP 的抑制。使用 Envision Multilabel Reader (PerkinElmer) 在 620 和 665 nm 处测量荧光。 FRET 通过 665 nm / 620 nm 的比率计算。 CHO-hMOR 细胞的平均载体比率为 3134 ± 99,DAMGO 的平均载体倍数为 2.2 ± 0.04。 CHO-hDOR 细胞的平均载体比率为 2962 ± 181,SNC80 的平均载体倍数为 1.6 ± 0.04。 CHO-hKOR 细胞的平均载体比率为 2965 ± 153,U69,593 的平均载体倍数为 1.9 ± 0.12。
SR17018是一种G蛋白偏向性μOR激动剂,对μOR的选择性高于δ-阿片受体(δOR)和κ-阿片受体(κOR)[1] 在G蛋白激活实验([³⁵S]GTPγS结合法)中,SR17018的EC₅₀为31 nM,最大效应(Emax)约为完全激动剂DAMGO的80%[1] 在β-arrestin 2募集实验(PathHunter实验)中,SR17018的效能降低,EC₅₀为144 nM,Emax约为DAMGO的30%,导致偏向因子为17(偏向G蛋白信号传导)[1] 在表达μOR的CHO细胞中,SR17018以剂量依赖性方式抑制毛喉素刺激的cAMP积累(EC₅₀ = 42 nM),与Gαi/o通路激活一致[1] 在稳定表达μOR的人胚胎肾(HEK293)细胞中,SR17018诱导μOR内化的程度低于DAMGO;流式细胞术分析显示,1 μM浓度下内化率约为20%(DAMGO为80%)[1] 用SR17018(1 μM,30分钟)预处理表达μOR的细胞,不会显著使μOR介导的G蛋白信号传导脱敏,而DAMGO(1 μM,30分钟)会导致明显脱敏(剩余活性:SR17018约70% vs DAMGO约30%)[1] |
||
| 体内研究 (In Vivo) |
通过腹膜内给药测定 C57BL/6J 小鼠的药代动力学参数。通过标准离心技术产生血浆,产生约 10 μL 血浆并立即冷冻。为了收集大脑,通过颈脱位处死小鼠,分离大脑并在液氮中快速冷冻。使用LC(岛津)串联质谱仪(AB Sciex)使用多反应监测方法以正离子模式操作来测定药物水平(Brust等人,2016)。使用快速平衡透析 (RED) 装置 (ThermoFisher) 测定芬太尼和吗啡的血浆蛋白结合。对于 SR 化合物,制备血浆样品(0.5 mL,0.5 μM 测试化合物)并将 900 μL 转移至 2 mL 聚碳酸酯超速离心管中。使用 Beckman Coulter Optima Max 超速离心机(最大 130,000 RPM)将样品以 400,000 xg 离心两小时,TLA 120.2 转子保持在 25°C。离心后的样品分为三层。富含蛋白质的底层含有大部分白蛋白,并且很容易可视化。顶层不易辨别,但含有高浓度的脂蛋白。中间层(使用所述条件下表面以下 1-2 毫米)具有非常低的蛋白质浓度,可用于确定未结合药物的量。通过 LC-MS/MS 通过比较离心样品中层的化合物浓度与未进行离心的平行样品的浓度来确定未结合化合物的百分比。
在小鼠热板试验(热痛模型)中,皮下注射(s.c.)SR17018可剂量依赖性产生镇痛作用,ED₅₀为1.2 mg/kg;该镇痛作用可被μOR选择性拮抗剂纳洛酮(1 mg/kg,s.c.)阻断[1] 在小鼠甩尾试验(伤害性反射模型)中,SR17018(0.3–3 mg/kg,s.c.)产生剂量依赖性镇痛作用,最大效应与DAMGO(10 mg/kg,s.c.)相当[1] SR17018(3 mg/kg,s.c.)不会在小鼠中引起明显呼吸抑制(通过全身体积描记法测量),而DAMGO(10 mg/kg,s.c.)会使分钟通气量降低40%[1] 在小鼠条件性位置偏爱(CPP)实验(奖赏寻求行为模型)中,SR17018(1–10 mg/kg,s.c.)未产生明显CPP,而DAMGO(5 mg/kg,s.c.)可诱导强烈CPP[1] SR17018(3 mg/kg,s.c.)不会在小鼠中引起明显便秘(通过24小时粪便颗粒产量测量),而DAMGO(10 mg/kg,s.c.)会使粪便产量减少约60%[1] 在小鼠中慢性给予SR17018(3 mg/kg,s.c.,每日一次,连续7天),不会诱导其镇痛作用的耐受性(热板试验);相反,DAMGO(10 mg/kg,s.c.,每日一次,连续7天)表现出明显耐受性(ED₅₀增加约3倍)[1] 慢性给予SR17018后,用纳洛酮诱发戒断(10 mg/kg,s.c.),未在小鼠中观察到戒断症状(跳跃、爪震颤),而DAMGO处理的小鼠表现出强烈戒断反应[1] |
||
| 酶活实验 |
利用商业酶片段互补测定(β-半乳糖苷酶)来确定 βarrestin2 招募到人 MOR 中。在测量信号之前,将 U2OS-βarrestin-hMOR PathHunter 细胞以每孔 5,000 个细胞的密度置于 384 孔白壁测定微孔板 (Greiner Bio-One) 中的 Assay Complete Cell Placing 5 Reagent (DiscoveRx) 中提前 16-20 小时。在 37°C 下用浓度递增的测试化合物处理细胞 90 分钟后,使用 PathHunter 检测试剂盒和 β-半乳糖苷酶底物评估 βarrestin2 募集情况,以检测功能性 β-半乳糖苷。使用 SpectraMax M5e 酶标仪(Molecular Devices)测量由此产生的发光增加。对于 PathHunter U2OS OPRM1 β 抑制蛋白细胞,平均载体为 446 ± 25 RLU,而 DAMGO 的平均载体倍数为 36 ± 1。
饱和和竞争放射性配体结合[1] 如前所述(Groer等人,2011;Schmid等人,2013),对CHO-hMOR、CHO-hDOR和CHO-hKOR细胞系进行了受体结合测定。将细胞在血清中饥饿30分钟,收集细胞,用特氟纶在玻璃上匀浆,在含有(50 mM Tris-HCl,pH 7.4,100 mM NaCl,1 mM EDTA)的膜缓冲液中制备膜丸,然后在4°C下以20000 x g离心30分钟。将膜重新悬浮在测定缓冲液(10 mM Tris-HCl,pH 7.4,100 mM NaCl)中。在25°C下,用适当的放射性配体(MOR,3H-DAMGO;KOR,3H-U69593;DOR,3H-二丙诺啡)在10μg膜上进行结合反应(200μl体积)2小时。对于竞争实验,每种放射性配体的浓度约为1 nM(0.96-1.10 nM 3H-DAMGO;1.06-1.19 nM 3H-U69593;0.92-0.98 nM ~3H-二丙诺啡)。在10μM DAMGO(MOR)、10μM U69593(KOR)或10μM纳洛酮(DOR)存在下测定非特异性结合。通过在Brandel细胞采集器上用GF/B玻璃纤维滤板过滤来终止反应,该滤板已与0.1%聚乙烯亚胺预孵育。在TopCount NXT闪烁计数器上用Microscint计数放射性。饱和结合分析和特异性结合的双曲线拟合用于确定CHO细胞系的放射性配体结合亲和力和受体数量(hMOR,3H-DAMGO为1.02±0.10 nM,1.58±0.11 pmol/mg;hDOR,0.70±0.11 nM[3H]-二丙诺啡和1.46±0.26 pmol/mg;hKOR,1.07±0.01 nM[3H]-U69593和0.71±0.12 pmol/mg])。 35S-GTPγS与膜结合[1] 如前所述(Schmid等人,2013),在从成年雄性C57BL/6J和MOR-KO小鼠分离的CHO hMOR和CHO mMOR细胞和脑干制备的膜中测定了35S-GTPγS结合。CHO-hMOR和CHO-mMOR细胞膜,如上所述用GTPγS结合膜缓冲液(10 mM Tris-HCl,pH 7.4,100 mM NaCl,1 mM EDTA)收集和制备。在25°C下,在悬浮在含有50μM鸟苷-5“-二磷酸(GDP)和0.1 nM 35S-GTPγS的测定缓冲液(50 mM Tris-Cl,pH 7.4,100 mM NaCl,5 mM MgCl2,1 mM EDTA)中的10μg膜上进行反应(200μl体积)1小时。通过GF/B滤板过滤终止反应,并如上所述计算放射性。对于分离自C57BL/6J和MOR-KO小鼠的脑干上的[35S]-GTPγS结合,用多电子组织撕裂机均质化组织,并如上所述制备膜。在收获前,将含有2.5μg蛋白质、1 mM二硫苏糖醇(DTT)、20μM GPD和0.1 nM 35S-GTPγS的结合反应在室温下孵育2小时。CHO hMOR膜的平均载体值为786±78cpm,DAMGO的平均折叠载体值为4.6±0.26。CHO mMOR细胞膜的平均载体值为694±28 cpm,DAMGO的平均折叠载体值为5.9±0.57。C57BL/6J脑干膜的平均载体为657±62 cpm,DAMGO的平均折叠载体为1.9±0.03。MOR-KO脑干膜的平均载体为1647±507cpm。 β受体阻滞2招募试验[1] 为了确定βarresti2对人MOR的募集,使用了商业酶片段互补分析(β-半乳糖苷酶)。在测量信号前16-20小时,将U2OS-βarrestin-hMOR PathHunter®细胞以每孔5000个细胞的密度铺在384孔、白壁的测定微孔板上的测定全细胞板5试剂中。在37°C下用浓度递增的测试化合物处理细胞90分钟,并使用带有β-半乳糖苷酶底物的PathHunter®检测试剂盒测定βarresti2的募集,以检测功能性β-半乳糖。使用SpectraMax M5e微孔板读数器测量由此产生的发光增加。PathHunter U2OS OPRM1βarresten细胞的平均载体为446±25 RLU,DAMGO的平均折叠载体为36±1。 为了确定βarresti2对mMOR的募集,使用了基于成像的检测方法(Zhou等人,2013)。在分析前16-20小时,将U2OS-βarrestein2-GFP-mOR细胞以每孔5000个细胞的密度铺在384孔、黑壁、透明底部光学成像微孔板(Brooks)的正常培养基中。细胞被血清饥饿1小时,然后在37°C下用越来越高浓度的测试化合物处理20分钟。用含4%多聚甲醛(PFA)的Hoechst核染色剂以1:1000的稀释度固定细胞。在CellInsight CX5高含量筛查平台上使用20X物镜测量βArrestin 2易位。使用Cellomics的斑点检测生物应用对Puncte(归一化为赫斯特染色)进行定量。载体处理的U2OS-βarrestiin2-GFP-mMOR细胞的平均泪点/赫斯特比为2.2±0.54,DAMGO的平均载体倍数为61±13。 为了确定化合物是否具有NOP活性,在U2OS-Ango-hOPRL1-bla细胞中测定了βarresti2对受体的募集。在测定前16-20小时,将U2OS-Ango-hOPRL1-bla细胞以每孔10000个细胞的密度铺在384孔、黑壁、透明底部的测定板上,置于32μl的测定介质(DMEM+10%透析FBS、0.1 mM NEAA、25 mM HEPES和1%笔/链球菌)中。在37°C下用浓度逐渐增加的试验化合物处理细胞5小时。根据制造商的方案,使用LiveBLAzer FRET B/G装载试剂盒和溶液D测定NOP活化。使用SpectraMax M5e微孔板阅读器测量FRET信号(激发409nm,发射460nm和530nm)。经460/530比值载体处理的U2OS-Ango-hOPRL1-bla细胞的平均值为0.31±0.03,伤害性感受肽的平均倍数载体为7.6±0.68。 采用[³H]DAMGO作为放射性配体,进行放射性配体结合实验以测定SR17018对μOR的Ki值;将表达μOR的细胞膜制剂与不同浓度的SR17018和固定浓度的[³H]DAMGO(0.5 nM)在25°C孵育90分钟;通过过滤分离结合态和游离态配体,测量放射性强度,采用非线性回归计算Ki值[1] 开展[³⁵S]GTPγS结合实验评估G蛋白激活:将表达μOR的细胞膜与SR17018(10 pM–10 μM)和[³⁵S]GTPγS(0.1 nM)在GDP(10 μM)存在下于30°C孵育60分钟;通过GF/B滤膜过滤、洗涤,定量结合的放射性强度,确定EC₅₀和Emax[1] 采用PathHunter系统进行β-arrestin 2募集实验:在384孔板中接种稳定表达μOR(标记ProLink)和β-arrestin 2(标记Enzyme Acceptor)的HEK293细胞;用SR17018(10 pM–10 μM)处理90分钟(37°C);加入底物孵育60分钟,测量化学发光强度,计算EC₅₀和Emax[1] |
||
| 细胞实验 |
cAMP accumumlation assay [1]
将 CHO-hMOR、-hDOR 和 -hKOR 细胞以每孔 4,000 个细胞的密度接种在 384 孔、白壁、30 μl 体积微孔板(Greiner Bio-One)中含有 1% FBS 的 Opti-MEM 中。 ) 测定前四小时。将 20 μM 毛喉素、25 μM 4-(3-丁氧基-4-甲氧基苯甲基)咪唑烷-2-酮 (Ro-20-1724) 和浓度逐渐升高的测试化合物应用于细胞,在 25°C 下持续 30 分钟。接下来,我们使用Cisbio的均质时间分辨荧光共振能量转移(FRET)cAMP HiRange测定(Cisbio-62AM6PEC)来测量cAMP的抑制作用。使用 PerkinElmer Envision Multilabel Reader 测量 620 和 665 nm 处的荧光。 FRET 的计算公式为 665 nm / 620 nm。对于 CHO-hMOR 细胞,平均媒介物比率为 3134 ± 99,而对于 DAMGO,平均媒介物倍数为 2.2 ± 0.04。对于 CHO-hDOR 细胞,平均载体比率为 2962 ± 181,对于 SNC80,平均载体倍数为 1.6 ± 0.04。对于 CHO-hKOR 细胞,平均媒介物比率为 2965 ± 153,对于 U69,593,平均媒介物倍数为 1.9 ± 0.12。 测量表达μOR的CHO细胞中cAMP的积累:在96孔板中接种细胞,过夜培养,先用SR17018(10 pM至10 μM)预处理30分钟,然后加入福司可林(10 μM)15分钟;裂解细胞,使用竞争性ELISA试剂盒定量cAMP水平,以确定EC₅₀值[1] 通过流式细胞术评估μOR的内化: 用荧光标记的μOR特异性抗体标记表达μOR的HEK293细胞, 在37°C下用SR17018 (1 μM)或DAMGO (1 μM)处理30分钟; 洗涤细胞, 固定, 然后使用流式细胞术分析荧光强度, 以量化内化百分比[1] 评估受体脱敏作用: 将表达μOR的细胞预先处理30分钟,使用SR17018(1 μM)或DAMGO(1 μM),然后洗去,再用一个亚最大浓度的DAMGO(100 nM)刺激;测量【³⁵S】GTPγS结合,以评估剩余的G蛋白信号传导活性[1] |
||
| 动物实验 |
|
||
| 毒性/毒理 (Toxicokinetics/TK) |
SR17018在镇痛剂量下不会引起小鼠明显的呼吸抑制、便秘或寻求奖赏行为[1]
长期服用SR17018不会导致小鼠产生耐受性或戒断症状[1] |
||
| 参考文献 | |||
| 其他信息 |
偏向性激动剂被认为是一种区分G蛋白偶联受体(GPCR)靶点下游药物的有益反应和不良反应的方法。本文描述了一系列μ-阿片受体(MOR)选择性激动剂的结构特征,这些激动剂优先激活受体与G蛋白偶联或募集β-arrestin蛋白。通过比较各通路中MOR介导信号的相对偏向性,我们证明了一系列化合物在呼吸抑制和伤害感受治疗窗口之间存在很强的相关性,这些化合物涵盖了广泛的信号偏向性。我们发现,β-arrestin偏向性化合物(例如芬太尼)在低镇痛剂量下更容易诱发呼吸抑制,而G蛋白信号偏向性则拓宽了治疗窗口,使得在不引起呼吸抑制的情况下也能发挥镇痛作用。[1]
尽管存在这些局限性,但本研究仍然是首个系统性地评估一系列激动剂在多种信号通路检测中偏向性,并以剂量依赖性方式对行为反应进行全面分析的研究。观察到的偏向性与治疗窗口宽度之间的相关性,对于利用这些信号通路检测来预测本研究中使用的鼠模型的疗效具有重要的参考价值。此外,我们还证明,可以通过对化学骨架的关键区域进行巧妙的修饰,来调控细胞培养中不同检测方法之间的信号传导,并且这种调控在体内也表现为效力差异。最后,本研究引入了一系列新的 G 蛋白信号偏向性 MOR 激动剂,这些激动剂在啮齿动物模型中实现了迄今为止报道的呼吸抑制和镇痛作用之间最高的分离度。我们希望这项研究能够帮助开发出比现有阿片类药物更安全的替代疗法。[1] SR17018 是一种合成的 G 蛋白偏向性 μ 阿片受体激动剂,旨在将镇痛作用与阿片类药物相关的不良反应(呼吸抑制、成瘾、便秘)区分开来。[1] 它对 G 蛋白信号传导的偏向性(而非 β-arrestin 2 募集)与受体内化、脱敏和耐受性的降低以及不良反应的减少有关。[1] SR17018 的偏向因子 (17) 与较宽的治疗窗相关,这体现在镇痛疗效和不良反应阈值之间的分离上。[1] SR17018 提供的临床前证据表明,具有高 G 蛋白偏向性的 μ 阿片受体激动剂可以作为更安全的阿片类镇痛药,降低滥用风险和副作用。 [1] |
| 分子式 |
C₁₉H₁₈CL₃N₃O
|
|
|---|---|---|
| 分子量 |
410.72
|
|
| 精确质量 |
409.05
|
|
| 元素分析 |
C, 55.56; H, 4.42; Cl, 25.89; N, 10.23; O, 3.90
|
|
| CAS号 |
2134602-45-0
|
|
| 相关CAS号 |
|
|
| PubChem CID |
130431397
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| LogP |
4.7
|
|
| tPSA |
35.6
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
2
|
|
| 可旋转键数目(RBC) |
3
|
|
| 重原子数目 |
26
|
|
| 分子复杂度/Complexity |
506
|
|
| 定义原子立体中心数目 |
0
|
|
| InChi Key |
LAGUDYUGRSQDKS-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C19H18Cl3N3O/c20-13-3-1-12(2-4-13)11-24-7-5-14(6-8-24)25-18-10-16(22)15(21)9-17(18)23-19(25)26/h1-4,9-10,14H,5-8,11H2,(H,23,26)
|
|
| 化学名 |
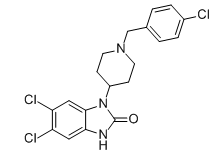
5,6-dichloro-3-[1-[(4-chlorophenyl)methyl]piperidin-4-yl]-1H-benzimidazol-2-one
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.25 mg/mL (3.04 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 12.5 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.25 mg/mL (3.04 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 12.5 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4347 mL | 12.1737 mL | 24.3475 mL | |
| 5 mM | 0.4869 mL | 2.4347 mL | 4.8695 mL | |
| 10 mM | 0.2435 mL | 1.2174 mL | 2.4347 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Functional Effects of MOR Agonists at the Human DOR, KOR, and NOP. |
|---|
SR Compounds Are Potent Activators of GTPγS Binding but Have Differential βARRESTIN2 Signaling Profiles at the Human MOR. |
GTPγS Binding at Mouse MOR Expressed in CHO Cells and Mouse Brainstem Compared to βArrestin2 Recruitment to Mouse MOR. |
SR Agonists Cross the Blood Brain Barrier and Are Present in Plasma 6 hr after Injection. |
|---|
Agonists That Displayed G Protein-Signaling Bias in the Cell-Based Assays Promote Antincocicpetion with Less Respiratory Suppression. |
Dose Response for the Fentanyl, Morphine, and the SR Compounds in the Antinociception and Respiratory Assays and Efficacy in Female Mice. |
 MOR modulator-1
MOR modulator-1
 Sunobinop TFA
Sunobinop TFA
 RM1490
RM1490
 Orphine
Orphine
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA






 463611831
463611831
