| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
M1 mAChR ( IC50 = 477 nM ); M1 mAChR ( Ki = 9.91 μM )
Muscarinic acetylcholine receptor M1 (M₁ mAChR): EC₅₀ = 198 ± 13.2 nM (calcium mobilization assay in CHO cells expressing rat M₁), AChₘₐₓ = 80.52% ± 7.67% max[2] - Muscarinic acetylcholine receptor M1 (M₁ mAChR): EC₅₀ = 379 ± 912 nM (calcium mobilization assay in rM₁ Y381A cells with orthosteric mutation)[2] - Muscarinic acetylcholine receptor M1 (M₁ mAChR): No agonism at M₂-M₅ mAChR subtypes up to 30 μM[2] - Muscarinic acetylcholine receptor M1 (M₁ mAChR): IC₅₀ > 30 μM for antagonism of ACh-induced responses at M₂-M₅ mAChR subtypes[2] - Muscarinic acetylcholine receptor M1 (M₁ mAChR): Does not compete with [³H]-NMS for binding to M₁-M₅ mAChRs (IC₅₀ > 30 μM)[2] - Muscarinic acetylcholine receptor M1 (M₁ mAChR): Shows agonist activity in calcium release assay (EC₅₀ not specified in this context) and ERK phosphorylation assay, but no effect on β-arrestin recruitment in hM₁ CHO cells[3] - Muscarinic acetylcholine receptor M1 (M₁ mAChR): At high concentrations, completely displaces [³H]-NMS binding to the orthosteric site of M₁-M₅ receptors (no specific IC₅₀ value provided)[1] |
|---|---|
| 体外研究 (In Vitro) |
体外活性:VU0357017 是一种 M1 选择性激动剂,似乎通过变构位点的作用激活 M1。来自竞争结合实验的 VU0357017 的 Ki 值分别为 9.91(rM1)、21.4 (rM2)、55.3 (rM3)、35 (rM4) 和 50 (rM5)。 VU0357017 是一种高度选择性的 M1 激动剂,表明这些化合物是不太可能在 M1 上高度保守的正构位点发挥作用,而更有可能充当变构激动剂。激酶测定:VU0357017 是一种 M1 选择性激动剂,似乎通过变构位点的作用激活 M1。来自竞争结合实验的VU0357017的Ki值分别为9.91(rM1)、21.4(rM2)、55.3(rM3)、35(rM4)和50(rM5)。细胞测定:
1. 在表达大鼠M₁ mAChR的CHO细胞钙动员实验中,VU0357017 HCI 表现出强效且高效的激动活性,EC₅₀为198 ± 13.2 nM,AChₘₐₓ为80.52% ± 7.67% max;在30 μM浓度下,对M₂-M₅ mAChR亚型无激动活性[2] 2. 在携带正构位点突变(Y381A,该突变可消除正构激动剂结合)的rM₁细胞中,VU0357017 HCI 仍能诱导功能性反应,EC₅₀为379 ± 912 nM,证实其变构激动剂作用机制[2] 3. 在评估对M₂-M₅亚型ACh诱导的EC₈₀反应拮抗作用的实验中,VU0357017 HCI 对所有M₂-M₅亚型的IC₅₀均>30 μM,显示出对M₁的高选择性[2] 4. 在M₁-M₅亚型的[³H]-NMS竞争结合实验中,VU0357017 HCI 的IC₅₀>30 μM,不与正构配体[³H]-NMS竞争结合,进一步支持其变构结合模式[2] 5. 在瞬时转染M₁受体突变体(E401H单点突变)的CHO-K1细胞中,VU0357017 HCI 效力显著下降(无具体EC₅₀值);而K392D、E397V或E397D突变对其效力影响较小,提示第三胞外环的E401是其结合的关键位点[2] 6. 在表达M₁-M₅受体的CHO细胞膜制备物中,高浓度VU0357017 HCI 可完全置换正构位点的[³H]-NMS结合;利用可诱导表达M₁的细胞系进行Furchgott分析显示,其为M₁的弱正构部分激动剂,且能减慢[³H]-NMS从CHO-rM₁细胞膜的解离速率,提示双位点(正构+变构)结合模式[1] 7. 在稳定表达人M₁ mAChR的CHO-K1细胞中,VU0357017 HCI 可诱导钙释放(以卡巴胆碱最大反应为基准归一化)和ERK1/2磷酸化(以基础ERK水平为基准的倍数变化),但在采用PathHunter检测试剂盒的β-arrestin募集实验中无活性(以% CCh max归一化)[3] 8. 在经不同浓度四环素处理(调控M₁受体密度)的hM₁ TREx CHO细胞中,VU0357017 HCI 以浓度依赖方式诱导钙释放(以% ionomycin归一化);50 ng/ml或1 μg/ml四环素诱导M₁表达后,对其在ERK磷酸化实验中的最大反应影响较小[3] 9. 在表达β-arrestin2-YFP的hM₁ TREx CHO细胞中,100 μM VU0357017 HCI 处理(无论细胞是否经50 ng/ml或1 μg/ml四环素预处理)均未诱导β-arrestin2募集[3] 10. 在大鼠海马脑片中,500 nM VU0357017 HCI 浴槽给药10分钟后进行阈下θ-爆发刺激(TBS),可显著增强Schaffer侧支-CA1突触的场兴奋性突触后电位(fEPSP)斜率,提升阈下θ-爆发长时程增强(LTP);30 μM VU0357017 HCI 浴槽给药10分钟对该突触的长时程抑制(LTD)无影响[3] 11. 在大鼠纹状体中型多棘神经元(MSNs)中,VU0357017 HCI 可诱导诱发动作电位发放频率小幅但显著升高(p ≤ 0.001),但作用弱于10 μM卡巴胆碱(CCh)[3] 12. 在小鼠内侧前额叶皮质(mPFC)锥体细胞中,VU0357017 HCI 无可检测的激动活性,也不影响自发性兴奋性突触后电流(sEPSCs)[3] |
| 体内研究 (In Vivo) |
VU0357017 在改善大鼠海马依赖性学习方面具有强大的功效。 VU0357017 增强大鼠在莫里斯水迷宫和情境恐惧调节中的表现。
1. 在啮齿类情境恐惧条件反射模型中,VU0357017 HCI(10 mg/kg,腹腔注射)可显著逆转东莨菪碱(0.2 mg/kg,皮下注射)诱导的情境恐惧条件反射获得障碍,表现为模型动物在训练环境中的僵住时间占比增加(与溶媒/东莨菪碱组相比p < 0.05)[2] 2. 在大鼠莫里斯水迷宫(MWM)实验中,VU0357017 HCI 处理可改善空间记忆表现(5天测试的游泳距离合并至每日四次试验分析);但在第4、5天的空间记忆测试(p > 0.445)、第4天第4次试验与第5天第1次试验间的记忆保持(p > 0.755)、以及探针试验前30秒的平台穿越次数(p > 0.981)中无显著效应[3] 3. 在大鼠情境恐惧条件反射(CFC)实验中,VU0357017 HCI 处理可增强情境恐惧的获得(与溶媒组相比p < 0.05)[3] 4. 在大鼠安非他命诱导的多动模型中,VU0357017 HCI(安非他命4 mg/kg皮下注射前30分钟腹腔给药)无法逆转安非他命诱导的多动行为,提示其无抗精神病样作用[3] |
| 酶活实验 |
1. M₁-M₅ mAChR的[³H]-NMS竞争结合实验:制备表达大鼠M₁-M₅ mAChR的CHO细胞膜,将其与[³H]-NMS(终浓度0.3 nM,溶于含100 mM NaCl和20 mM HEPES的缓冲液,pH=7.4)及不同浓度的VU0357017 HCI 或阿托品(阳性对照)在室温下孵育3小时。通过快速过滤终止平衡结合反应,以特异性[³H]-NMS结合率的百分比绘制数据曲线。实验证实VU0357017 HCI 不与[³H]-NMS竞争结合M₁-M₅ mAChR(IC₅₀>30 μM)[2]
2. M₁ mAChR的[³H]-NMS解离实验:制备稳定表达大鼠M₁受体的CHO细胞膜,与[³H]-NMS孵育至平衡结合后,加入阿托品诱导[³H]-NMS解离,检测10 μM或1 mM VU0357017 HCI 对解离速率(k_off)的影响。结果显示VU0357017 HCI 可减慢[³H]-NMS解离速率(10 μM时k_off=0.030 ± 0.006/min;1 mM时k_off=0.025 ± 0.006/min),而阿托品单独处理组k_off=0.041 ± 0.001/min,提示其通过变构位点与正构配体产生负协同作用[1] |
| 细胞实验 |
1. M₁ mAChR激动活性的钙动员实验:将表达大鼠M₁-M₅ mAChR的CHO细胞接种至合适孔板,负载钙敏感荧光染料,孵育后加入不同浓度的VU0357017 HCI,利用荧光酶标仪检测细胞内钙浓度变化,绘制浓度-反应曲线并计算EC₅₀和最大效应(AChₘₐₓ)。实验证实VU0357017 HCI 是强效M₁激动剂,在30 μM浓度下对M₂-M₅无活性[2]
2. M₁受体突变体细胞的钙动员实验:将瞬时转染M₁受体突变体(E401H、K392D、E397V等)的CHO-K1细胞按上述钙动员实验流程处理,加入不同浓度的VU0357017 HCI 并测定EC₅₀,分析突变位点对其效力的影响[2] 3. ERK1/2磷酸化实验:将hM₁ CHO细胞或经不同浓度四环素处理(调控M₁表达)的hM₁ TREx CHO细胞用不同浓度VU0357017 HCI 处理,裂解细胞后采用SureFire ERK磷酸化试剂盒检测ERK1/2磷酸化水平,数据以基础ERK水平的倍数变化表示,并以卡巴胆碱最大反应为基准归一化[3] 4. β-arrestin募集实验:采用PathHunter检测试剂盒,在hM₁ CHO细胞中加入不同浓度VU0357017 HCI,检测β-arrestin募集并以% CCh max归一化;同时,在表达β-arrestin2-YFP的hM₁ TREx CHO细胞中加入100 μM VU0357017 HCI,通过共聚焦显微镜观察并计数斑点数量以量化β-arrestin2募集[3] 5. 可诱导M₁细胞系的Furchgott分析:将hM₁ TREx CHO细胞用1 μg/mL或25 ng/mL四环素处理24小时以调控M₁受体密度,在钙释放实验中绘制VU0357017 HCI 的浓度-反应曲线,通过等效应浓度的双倒数图计算平衡解离常数(pK_d和K_d),证实其为M₁的弱正构部分激动剂[1] |
| 动物实验 |
雄性Sprague-Dawley大鼠(380-420 g)预先接受东莨菪碱处理
1、3、10 mg/kg 单次腹腔注射 1. 啮齿动物情境恐惧条件反射实验:啮齿动物预先接受东莨菪碱(0.2 mg/kg,皮下注射)处理以诱导认知功能障碍。将VU0357017 HCI溶解于适当的溶剂(未具体说明)中,并以10 mg/kg的剂量腹腔注射给药。然后对动物进行情境恐惧条件反射训练(暴露于特定情境并伴有轻微足底电击)。经过一段保持期后,测量动物在同一情境中僵住不动的时间百分比,以评估其认知功能; VU0357017 HCI 逆转了东莨菪碱诱导的缺陷(每组治疗组 n = 3-4 只大鼠)[2] 2. 大鼠莫里斯水迷宫 (MWM) 实验:将 VU0357017 HCI 溶解于合适的溶剂(未指定)中,并通过未指定的途径(频率未指定)给予大鼠(每组 n = 8 只)。大鼠在 MWM 中训练 5 天(每天 4 次试验),以找到隐藏的平台;每日记录游泳距离。在第 5 天进行探针试验(移除平台),并计数前 30 秒内的平台穿越次数,以评估空间记忆保持情况[3] 3. 大鼠情境恐惧条件反射 (CFC) 实验:将 VU0357017 HCI 溶解于合适的溶剂(未指定)中,并通过未指定的途径给予大鼠(每组 n = 4-6 只)。大鼠接受条件性恐惧训练(暴露于特定环境并接受足底电击),训练后测量其僵直行为的百分比,以评估其对环境恐惧的习得情况[3] 4. 大鼠安非他明诱导的运动过度试验:将VU0357017 HCI溶解于溶剂(未具体说明)中,并在皮下注射安非他明(4 mg/kg)前30分钟腹腔注射。将大鼠置于开放式场地中,并在特定时间内测量其运动活性(运动距离),以评估VU0357017 HCI逆转安非他明诱导的运动过度的能力[3] 5. 大鼠海马切片的电生理研究:制备大鼠海马切片,并将其保存在32°C的人工脑脊液(ACSF)中。将VU0357017 HCl溶解于人工脑脊液(ACSF)中,并在刺激施加前10分钟以500 nM(用于长时程增强(LTP)研究)或30 μM(用于长时程抑制(LTD)研究)的浓度进行灌流。在Schaffer侧支-CA1突触处记录场兴奋性突触后电位(fEPSP)以评估LTP和LTD[3] 6. 大鼠纹状体中型棘状神经元(MSN)的电生理记录:制备大鼠纹状体切片,并对MSN进行全细胞膜片钳记录。 VU0357017 HCI 通过灌流方式施加,并测量电流阶跃刺激下的动作电位发放频率,以评估其对神经元兴奋性的影响[3] 7. 小鼠内侧前额叶皮层 (mPFC) 锥体细胞的电生理记录:制备小鼠 mPFC 切片,并对锥体细胞进行全细胞膜片钳记录。VU0357017 HCI 通过灌流方式施加,并记录自发性兴奋性突触后电流 (sEPSC),以评估其激动剂活性[3] |
| 药代性质 (ADME/PK) |
VU0357017 HCI 在全身给药后可提供良好的脑暴露量(未提供具体的药代动力学参数,例如半衰期、口服生物利用度或清除率)[2]
|
| 参考文献 |
|
| 其他信息 |
1. VU0357017 HCI 是一种高选择性的 M₁ 毒蕈碱乙酰胆碱受体变构激动剂,是通过功能性高通量筛选 (HTS) 和多样性导向合成方法发现的;它具有前所未有的纯净辅助药理学特性,在 10 μM 浓度下对大量靶点均无明显活性[2]
2. 靶向诱变研究表明,M₁ 受体的第三个胞外环(尤其是 E401)和跨膜区第七个螺旋圈对于 VU0357017 HCI 的结合至关重要;柔性配体对接实验表明,VU0357017 HCI 可与 M₁ 受体的 E401、E397、Y381 和 Q181 形成多达四个氢键[2] 3. VU0357017 HCI 作为 M₁ mAChR 的双位点配体,具有弱的正构部分激动剂活性和变构相互作用(减缓 [³H]-NMS 的解离),这使其区别于纯变构激动剂[1] 4. VU0357017 HCI 差异性地激活 M₁ 与不同信号通路的偶联(诱导钙释放和 ERK 磷酸化,但不募集 β-arrestin),从而对大脑回路产生选择性作用:在海马体中具有显著作用(增强 LTP 和认知功能),在纹状体中作用较弱,而在内侧前额叶皮层中无作用[3] 5. VU0357017 HCI是研究M₁介导的中枢神经系统(CNS)效应的宝贵研究工具,也是阿尔茨海默病和精神分裂症新疗法的潜在先导化合物[2] |
| 分子式 |
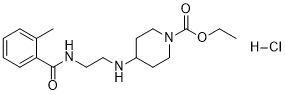
C18H28CLN3O3
|
|
|---|---|---|
| 分子量 |
369.89
|
|
| 精确质量 |
369.182
|
|
| 元素分析 |
C, 58.45; H, 7.63; Cl, 9.58; N, 11.36; O, 12.98
|
|
| CAS号 |
1135242-13-5
|
|
| 相关CAS号 |
|
|
| PubChem CID |
25010775
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| LogP |
3.64
|
|
| tPSA |
74.16
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
25
|
|
| 分子复杂度/Complexity |
408
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
Cl[H].O(C([H])([H])C([H])([H])[H])C(N1C([H])([H])C([H])([H])C([H])(C([H])([H])C1([H])[H])N([H])C([H])([H])C([H])([H])N([H])C(C1=C([H])C([H])=C([H])C([H])=C1C([H])([H])[H])=O)=O
|
|
| InChi Key |
XKJQVUIXSBOCPP-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C18H27N3O3.ClH/c1-3-24-18(23)21-12-8-15(9-13-21)19-10-11-20-17(22)16-7-5-4-6-14(16)2;/h4-7,15,19H,3,8-13H2,1-2H3,(H,20,22);1H
|
|
| 化学名 |
ethyl 4-[2-[(2-methylbenzoyl)amino]ethylamino]piperidine-1-carboxylate;hydrochloride
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 25~38 mg/mL (67.6~102.7 mM)
Water: ~10 mg/mL Ethanol: ~2 mg/mL |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.76 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.76 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.76 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 33.33 mg/mL (90.11 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7035 mL | 13.5175 mL | 27.0351 mL | |
| 5 mM | 0.5407 mL | 2.7035 mL | 5.4070 mL | |
| 10 mM | 0.2704 mL | 1.3518 mL | 2.7035 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
