| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 100mg |
|

| 靶点 |
wild-type ROS1; ROS1 G2032R; ROS1 fusions and resistance mutants
Zidesamtinib targets ROS1 (ROS proto-oncogene 1, receptor tyrosine kinase), a receptor tyrosine kinase that is involved in cell proliferation and survival. Fusions of the ROS1 gene lead to constitutive activation of the kinase and are oncogenic drivers in a subset of cancers, most notably NSCLC. Zidesamtinib potently inhibits ROS1 with an IC50 of 0.7 nM. It also inhibits the ROS1 G2032R resistance mutation, which is a common mechanism of acquired resistance to first-generation ROS1 inhibitors. |
|---|---|
| 体外研究 (In Vitro) |
齐德沙替尼(72 小时)平均 IC50 值为 0.4 nM,可抑制七种表达野生型 ROS1 融合蛋白的细胞系的生长[1]。
齐德沙替尼(72 小时)平均 IC50 值为 1.6 nM,可抑制六种携带 G2032R 突变 ROS1 融合蛋白的细胞系的生长[1]。 齐德沙替尼(72 小时)可有效抑制非 G2032R ROS1 突变体,IC50 值 ≤ 1.5 nM[1]。 齐德沙替尼(10-1000 nM;4 周)可抑制表达 G2032R 突变 ROS1 融合蛋白和野生型 ROS1 融合蛋白的 NIH3T3 细胞的克隆形成[1]。 体外实验表明,齐德沙替尼是一种高效且选择性的 ROS1 抑制剂。对野生型 ROS1 的 IC50 值为 0.7 nM。它对 ROS1 G2032R 突变体和其他耐药突变体均有活性。与其他激酶相比,它对 ROS1 具有高度选择性,从而最大限度地减少了脱靶效应。其能够穿过血脑屏障的能力是它区别于其他 ROS1 抑制剂的关键特征。 |
| 体内研究 (In Vivo) |
齐德沙替尼(0.04-15 mg/kg;口服,每日两次,持续28天)在所有剂量≥0.2 mg/kg时均可诱导野生型ROS1异种移植瘤模型的肿瘤消退[1]。体内研究表明,齐德沙替尼在临床前模型中具有强大的抗肿瘤活性,包括脑转移瘤模型。动物实验中,齐德沙替尼的给药剂量范围为0.04至15 mg/kg,均采用灌胃给药。其良好的脑渗透性使其能够在中枢神经系统中达到治疗浓度,这对于治疗脑转移瘤患者至关重要。
|
| 酶活实验 |
生化激酶活性测定[1]
使用 PhosphoSens 测定法测定纯化激酶的活性。将测试化合物溶解于 DMSO 中,浓度为所需浓度的 100 倍,并以 250 nL 的量分装到 384 孔板中,进行 3 倍稀释系列实验。向孔板中加入 12.5 μL 含有 2 mmol/L ATP 和 26 μmol/L 荧光肽底物 AQT0101 或 AQT0104 的缓冲液(50 mmol/L HEPES pH 7.5、0.01% Brij-35、0.5 mmol/L EGTA、10 mmol/L MgCl2)。通过加入含有 0.5 nmol/L ROS1、2 nmol/L ROS1 G2032R、1 nmol/L TRKA、3 nmol/L TRKB 或 0.5 nmol/L TRKC 激酶结构域的 12.5 μL 溶液来引发反应,该溶液溶于缓冲液(50 mmol/L HEPES pH 7.5、0.01% Brij-35、2% 甘油、0.4 mg/mL BSA、0.5 mmol/L EGTA、10 mmol/L MgCl2)中。最终浓度为:1 mmol/L ATP,13 μmol/L 肽底物(ROS1 和 ROS1 G2032R 使用 AQT0101,TRKA、TRKB 和 TRKC 使用 AQT0104),0.25 至 1.5 nmol/L 激酶(0.25 nmol/L ROS1;1 nmol/L ROS1 G2032R;0.5 nmol/L TRKA;1.5 nmol/L TRKB;或 0.5 nmol/L TRKC),50 mmol/L HEPES pH 7.5,0.01% Brij-35,1% 甘油,0.2 mg/mL BSA,0.5 mmol/L EGTA,以及 10 mmol/L MgCl2。将反应板密封后,使用酶标仪在 λemission = 485 nm 波长下,于 30°C 下每 2 分钟记录一次荧光信号,持续 120 分钟。反应初始线性阶段荧光强度随时间的变化即为初始反应速率 (v)。IC50 值通过绘制初始反应速率与 log(抑制剂浓度) 的关系图,并回归四参数逻辑方程计算得出。[1] 激酶筛选[1] 使用放射性标记的 [γ-33P]-ATP 测定激酶活性抑制情况。将含有 [γ-33P]-ATP 的溶液与 NVL-520、335 种激酶及其相应的激酶底物混合。NVL-520 的测定浓度为 100 nmol/L 和 1 μmol/L。该测定中 ATP 的浓度接近每种激酶的米氏常数 (KM)。反应进行1小时后终止,并使用闪烁计数器定量测定33P的掺入量。抑制率通过33P的残余活性来衡量:残余活性越低,表明对特定激酶的抑制效力越高。基于335种激酶的筛选,选择24种抑制率>50%的激酶,使用相同的检测方法,在10个NVL-520浓度下(3 μmol/L、900 nmol/L、300 nmol/L、90 nmol/L、…、0.09 nmol/L)进行IC50的测定。IC50值通过绘制残余活性与log(抑制剂浓度)的关系图,并回归到四参数逻辑方程来确定。激酶组图谱由Cell Signaling Technology公司提供。 齐德沙替尼的抑制活性通过体外激酶活性测定进行评估。将重组ROS1激酶(野生型或突变型)与底物和不同浓度的化合物一起孵育。根据剂量反应曲线计算IC50值。通过筛选一系列其他激酶来验证其选择性。可以使用体外模型或通过测量体内脑血浆比来评估化合物穿过血脑屏障的能力。 |
| 细胞实验 |
细胞活力检测[1]
不同检测点的实验方案略有不同。在检测点 A,将 A549 或稳定 Ba/F3 细胞接种于 384 孔板中,并在含 10% FBS 的完全培养基中以 3 倍系列稀释加入待测化合物。与抑制剂孵育 72 小时后,使用 CellTiter-Glo 试剂盒检测细胞活力。未处理的孔作为阴性对照(增殖未受抑制),而用高浓度非特异性激酶抑制剂星形孢菌素处理的孔作为阳性对照(增殖完全抑制)。IC50 值由抑制率和抑制剂浓度的对数通过四参数逻辑回归计算得出。[1] 在检测点 B,所有抑制剂均配制成 1 mmol/L 的 DMSO 储备液。使用 Multidrop Combi 试剂分液器,每孔预先加入 25 μL 完全培养基。使用D300数字分液器,将抑制剂以指定浓度的2倍稀释至384孔板中,每孔加入25 μL完全培养基。使用Multidrop Combi试剂分液器,将表达野生型或突变型ROS1融合蛋白的Ba/F3细胞系以每孔1000个细胞的密度接种于25 μL培养基中。培养72小时后,采用WST-8 [2-(2-甲氧基-4-硝基苯基)-3-(4-硝基苯基)-5-(2,4-二磺基苯基)-2H-四唑单钠盐]法测定细胞活力,并在Biotek Synergy 2酶标仪上读取结果。每种条件均进行三次重复实验。使用 Microsoft Excel 对数据进行标准化,并使用 GraphPad Prism 中的非线性回归分析计算 IC50 值。[1] 在 C 位点测试中,将 MGH193-1 和 MGH9018-1 细胞以 4,000 个细胞/孔的密度接种于 96 孔板中,每个处理设置三个复孔。药物处理 5 天后,用 CellTiter-Glo 孵育细胞,并使用 SpectraMax M5 多功能微孔板读数仪测量发光值。使用 GraphPad Prism(GraphPad Software)以图形方式显示数据,并通过采用四参数分析方法的非线性回归模型确定 IC50 值。[1] 在某些人类癌细胞系中,我们观察到 NVL-520 表现出双相剂量反应行为。我们将第一个剂量反应归因于 ROS1 抑制引起的靶向生长抑制,而第二个剂量反应归因于 ROS1 抑制以外的通路引起的脱靶细胞毒性。在这种情况下,仅报告第一个 IC50 值,表明 ROS1 靶向抑制。[1] 细胞磷酸化分析[1] 对于 Ba/F3 TRKB 细胞磷酸化分析,将细胞接种于 384 孔板中,并在含 10% FBS 的完全培养基中以 3 倍稀释系列加入测试化合物。用 100 ng/mL BDNF 刺激细胞 20 分钟。使用磷酸化 TRKA (Tyr674/675)/磷酸化 TRKB (Tyr706/Tyr707) AlphaLISA 试剂测定 TRK 磷酸化水平。未处理的孔作为阴性对照(无抑制),而用高浓度非特异性激酶抑制剂星形孢菌素处理的孔作为阳性对照(完全抑制)。 IC50值是根据抑制率和抑制剂浓度,利用四参数逻辑回归计算得出的。[1] 表达EZR-ROS1野生型或突变型融合蛋白的NIH3T3细胞在收获前用指定浓度的抑制剂处理3小时。细胞用磷酸盐缓冲液(PBS)洗涤,并用添加了0.25%脱氧胆酸钠、0.05% SDS以及蛋白酶和磷酸酶抑制剂的细胞裂解缓冲液进行收集。使用Pierce BCA蛋白测定法测定蛋白浓度。裂解液用添加了β-巯基乙醇的Laemelli样品缓冲液在75℃下提取10分钟,并在4%至20%预制梯度Bis-Tris凝胶上进行电泳。[1] MGH193-1和MGH9018-1细胞处理6小时。使用以下抗体通过蛋白质印迹法分析总蛋白裂解物:磷酸化 ROS1 Y2274 (3078)、ROS1 (3287)、磷酸化 AKT S473 (4060)、AKT (4691)、磷酸化 ERK1/2 T202/Y204 (9101)、ERK1/2 (9102)、磷酸化 S6 S240/244 (5364)、S6 (2217) 和 β-肌动蛋白 (4970)。[1] 集落形成实验[1] 将培养板预先接种 0.8% 琼脂糖的完全培养基,培养基中添加 DMSO 或抑制剂(克唑替尼、恩曲替尼、劳拉替尼或 NVL-520,浓度分别为 10、100 或 1,000 nmol/L)。每种抑制剂均与其对应的DMSO处理组配对,作为精确的对照。表达CD74-ROS1或EZR-ROS1野生型或突变型融合蛋白的NIH3T3细胞以每0.5 mL琼脂糖2000个细胞的密度接种于含0.4%琼脂糖的完全培养基中,并在琼脂糖层中加入与底层浓度相同的DMSO或抑制剂。培养4周后,每周向每个孔中加入75 μL完全培养基(含或不含抑制剂),以匹配每种接种条件,防止琼脂糖干燥。分别在3周和4周后使用GelCount软件读取培养板。将各条件下的菌落计数取平均值,并以配对的DMSO处理组的菌落计数进行标准化。数据分析和可视化使用Microsoft Excel和GraphPad Prism软件完成。 在ROS1依赖性癌细胞系中评估了zidesamtinib的细胞活性。用齐德沙替尼处理细胞,并通过蛋白质印迹法检测其对ROS1磷酸化及其下游信号通路(例如ERK、AKT)的影响。采用标准方法评估其对细胞增殖和活力的影响。在表达突变型ROS1的细胞中证实了该化合物对耐药突变的活性。 |
| 动物实验 |
将CTG-0848模型[1]的肿瘤碎片皮下植入雌性无胸腺裸鼠(Nude-Foxn1nu)体内。剂量分别为0.04、0.2、1、5和15 mg/kg,每日两次灌胃,持续21天。皮下异种移植研究[1]:CTG-0848 PDX。[1]实验操作均按照Champions Oncology机构动物护理和使用委员会(IACUC)批准的指南进行,该机构已获得实验动物护理评估和认证协会(AAALAC)的认证。将CTG-0848模型的肿瘤碎片皮下植入雌性无胸腺裸鼠(Nude-Foxn1nu)左侧腹部。在疗效研究中,当肿瘤生长至 150 至 300 mm³ 后,将小鼠(每组 n = 3–5 只)随机分组,并通过灌胃法每日两次(间隔 12 小时)分别给予赋形剂或 NVL-520。在另一项药代动力学 (PK) 和药效学 (PD) 研究中,当肿瘤生长至 350 至 500 mm³ 后,小鼠接受单次给药或每日两次(连续 5 天)的赋形剂或 NVL-520 给药,并在给药后 1 小时和 12 小时(仅治疗组)采集肿瘤组织和血液样本。经供应商提供的NGS验证,该模型包含杂合CD74-ROS1融合基因。[1]
查看更多Lu01-0414 PDX。[1] CTG-2532 PDX。[1] 实验操作均按照经AAALAC认证的Champions Oncology机构动物护理与使用委员会(IACUC)批准的指南进行。将CTG-2532模型肿瘤组织块皮下植入雌性无胸腺裸鼠(Nude-Foxn1nu)左侧腹部。在疗效研究中,当肿瘤生长至150至300 mm³后,若小鼠耐受良好,则分别给予赋形剂、NVL-520或repotrectinib,每日两次(间隔12小时)灌胃给药,持续21天(每日两次,共21天)。瑞波替尼(DC Chemicals)以含有0.5%羧甲基纤维素和1%吐温-80的悬浮液形式给药。给药悬浮液在2–8°C避光条件下持续搅拌保存,最长可达7天。在另一项药代动力学(PK)和药效学(PD)研究中,将CTG-2532肿瘤皮下植入小鼠体内,待肿瘤生长至350至550 mm³后,给予单剂量载体或NVL-520。分别于给药后1小时和12小时(仅治疗组)采集肿瘤和血液样本,用于PK和PD分析。该模型经供应商提供的NGS验证,含有CD74–ROS1 G2032R.Ba/F3 CD74–ROS1 G2032R CDX。本研究中所有与动物操作、护理和治疗相关的程序均按照Pharmaron公司IACUC批准的指南,并遵循AAALAC的指导进行。将 1 × 10⁶ 个 Ba/F3 CD74–ROS1 G2032R 细胞皮下接种到 6 至 8 周龄的雌性 Balb/c 裸鼠右侧腹部。当肿瘤平均体积达到约 128 mm³ 时,开始治疗,并定期测量肿瘤和体重。[1] 采用液相色谱-质谱联用 (LC/MS) 法测定血浆药物浓度。游离药物浓度通过将药代动力学实验测定的总浓度值乘以小鼠血浆中相应的游离分数(NVL-520 为 7.7%,克唑替尼为 7.6%)计算得出。[1] Kp,uu 和颅内研究[1] 本研究中所有与动物操作、护理和治疗相关的程序均按照 Pharmaron 公司 IACUC 批准的指南执行,并遵循 AAALAC 的指导原则。[1] NVL-520 配制成 1 mg/mL 的 20% HP-β-CD 去离子水混悬液。劳拉替尼配制成 1 mg/mL 的 HCl + 20% HP-β-CD 去离子水溶液。将化合物口服给予雄性 Wistar Han 大鼠(每组 n = 3)。1 小时后,收集脑组织样本和血浆,并将脑组织样本在 PBS 中匀浆。脑组织和血浆样本经乙腈沉淀和离心(4700 rpm,15 分钟)后,采用液相色谱-串联质谱法(LC/MS-MS)定量上清液中的药物浓度。游离药物浓度采用快速平衡透析法测定。Kp,uu 计算为脑组织中游离药物浓度与血浆中游离药物浓度的比值。[1] Ba/F3 CD74–ROS1 G2032R 细胞经含有萤火虫荧光素酶基因和新霉素抗性标记的病毒颗粒转导。感染细胞经新霉素筛选,并通过有限稀释法建立单克隆培养。转化成功的细胞经 Sanger 测序和生物发光法验证。在体内研究中,将 1 × 10⁵ 个 Ba/F3 CD74–ROS1 G2032R 荧光素酶细胞立体定位植入 6 至 8 周龄雌性 Balb/c 裸鼠的右侧前脑。5 天后,根据平均生物发光信号将小鼠随机分为三组,每组 n = 7–10 只小鼠,分别接受载体或 NVL-520(2 mg/kg,每日两次)口服给药。定期测量生物发光和体重,直至研究结束(治疗开始后 61 天)或动物达到安乐死标准。在动物研究中,zidesamtinib 通常口服给药于携带皮下或颅内肿瘤异种移植瘤的啮齿动物。剂量范围为 0.04 至 15 mg/kg。检测肿瘤生长抑制情况,并评估药效学标志物,例如肿瘤组织中的ROS1磷酸化水平。通过测量脑组织和血浆中的药物浓度来确认药物的脑渗透性。 |
| 药代性质 (ADME/PK) |
作为一种口服活性小分子药物,齐德沙替尼的设计旨在方便给药。其能够穿过血脑屏障是其关键的药代动力学特征。临床前和临床研究提供了详细的药代动力学参数,例如半衰期和生物利用度。
|
| 毒性/毒理 (Toxicokinetics/TK) |
公开资料中关于齐德沙替尼的毒理学数据并不详尽。作为一种靶向治疗药物,其安全性预计可控。该药物对ROS1的选择性优于其他激酶,旨在最大限度地减少脱靶毒性。目前正在进行的临床试验中对其安全性进行评估。
|
| 参考文献 | |
| 其他信息 |
齐德沙替尼是一种口服的选择性受体酪氨酸激酶c-ros癌基因1 (ROS1) 抑制剂,具有潜在的抗肿瘤活性。口服后,齐德沙替尼靶向并结合ROS1的野生型、点突变型和融合蛋白,从而抑制ROS1驱动的肿瘤细胞增殖,包括携带某些ROS1耐药突变的肿瘤细胞,例如溶剂前沿突变G2032R以及S1986Y/F、L2026M和D2033N耐药突变。抑制ROS1可阻断下游信号通路,并抑制ROS1过表达、重排或突变的肿瘤细胞的生长。ROS1在某些癌细胞中过表达,并在癌细胞的生长和存活中起着至关重要的作用。NVL-520可以穿过血脑屏障(BBB)。
NVL-520 的全面临床前特征包括:对 ROS1 及其多种耐药突变具有强效靶向性,对 ROS1 G2032R 相对于 TRK 具有高选择性,以及能够穿过血脑屏障。这些特征标志着一种独特的 ROS1 TKI 的诞生,它有望克服早期 TKI 在 ROS1 融合阳性患者中的局限性。[1] Zidesamtinib(NVL-520,CAS:2739829-00-4)是一种旨在克服第一代抑制剂局限性的新一代 ROS1 抑制剂。它对耐药突变的强效活性以及穿过血脑屏障的能力使其成为 ROS1 阳性非小细胞肺癌(NSCLC)患者(包括脑转移患者)的一种极具前景的治疗药物。它的研发代表了 ROS1 驱动型癌症靶向治疗领域的一项重大进展。 |
| 分子式 |
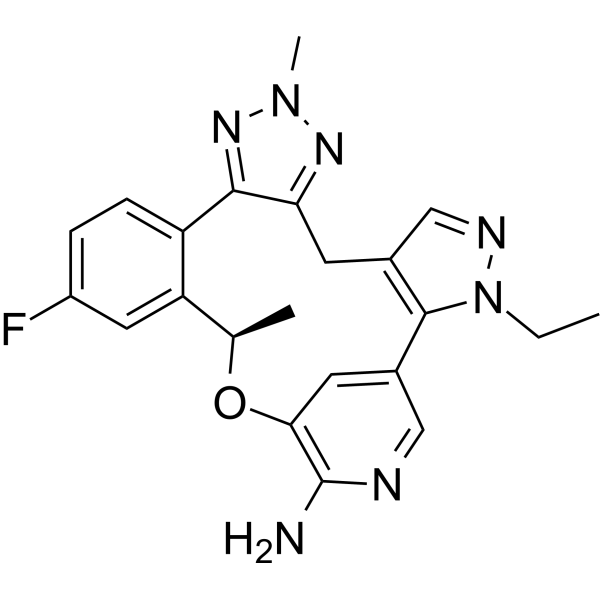
C22H22FN7O
|
|---|---|
| 精确质量 |
419.46
|
| 元素分析 |
C, 63.00; H, 5.29; F, 4.53; N, 23.37; O, 3.81
|
| CAS号 |
2739829-00-4
|
| 相关CAS号 |
2739829-00-4
|
| PubChem CID |
166560233
|
| 外观&性状 |
Typically exists as white to off-white solids at room temperature
|
| LogP |
2.8
|
| tPSA |
96.7Ų
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
1
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
629
|
| 定义原子立体中心数目 |
1
|
| SMILES |
CCN1C2=C(CC3=NN(N=C3C4=C(C=C(C=C4)F)[C@H](OC5=C(N=CC2=C5)N)C)C)C=N1
|
| InChi Key |
DTWUUAFTYSMNQX-GFCCVEGCSA-N
|
| InChi Code |
InChI=1S/C22H22FN7O/c1-4-30-21-13(11-26-30)7-18-20(28-29(3)27-18)16-6-5-15(23)9-17(16)12(2)31-19-8-14(21)10-25-22(19)24/h5-6,8-12H,4,7H2,1-3H3,(H2,24,25)/t12-/m1/s1
|
| 化学名 |
(19R)-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,9,10,11,23-hexazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine
|
| 别名 |
NVL520; NVL 520; NVL-520; Zidesamtinib; NVL-520; 2739829-00-4; NVL520; zidesamtinib [INN]; MX5KQV5XHC; Zidesamtinib [WHO-DD]; ZIDESAMTINIB [USAN];
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~119.20 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.25 mg/mL (2.98 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 12.5 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.25 mg/mL (2.98 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 12.5 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.25 mg/mL (2.98 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































 463611831
463611831
