| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
|
|
|---|---|---|
| 体外研究 (In Vitro) |
以剂量依赖性方式降低去饱和指数是 A939572 的强体内活性之一[1]。 SCD1 酶活性被一种名为 A939572 的微小化学物质选择性抑制。第 5 天,A939572 显示 Caki1、A498、Caki2 和 ACHN 增殖的统计学显着剂量依赖性降低(IC50 分别为 65 nM、50 nM、65 nM 和 6 nM)。与 DMSO+BSA 对照相比,A939572 (SCDi) 处理的 Caki1 和 A498 细胞中所有五种内质网应激相关基因表达水平均显着升高。通过添加 OA-BSA 可以防止这种增强的表达。
优化从检查苯氧环上的取代开始。对于邻位取代的类似物,IC50从H(4a,370 nM)和F(4b,98 nM)稳步下降到更大的卤素基团,如Cl和Br(4c/A939572和4d,<4 nM)。与哒嗪系列观察到的SAR趋势一致,苯氧基环上亲脂性增加,特别是邻位亲脂性,与抑制活性的提高相关。可以观察到,引入2-甲基导致抑制剂(4e,17 nM)的效力提高了20倍以上。相对亲水性更强的甲氧基类似物(4f,81 nM)的影响不太明显。如化合物4g(20 nM)所示,间位取代也具有良好的耐受性,尽管不如邻位取代的类似物4c/A939572(<4 nM)最佳。为了探索任何额外的亲脂性相互作用,我们在4c的基础上在4、5或6位引入了氟化物。如表1所示,2,4-二取代的类似物4j能够保持4c观察到的大部分效力,而2,6-二取代则放弃了2-单取代产生的所有增益(4h,330 nM vs 4a,370 nM)。发现在5位同时存在F是有益的,因为2,5-萘醌4i在人SCD1中表现出约两倍的效力提高(4i,hSCD1 IC50=18 nM vs 4c,hSCT1 IC50=37 nM)。[1] 基于与化合物4c/A939572相关的令人鼓舞的结果,我们接下来研究了用杂环部分取代酰胺基团以进一步提高SCD1抑制活性。这种修饰产生了几个IC50保留良好的类似物(4l,8 nM;4m,10 nM),尽管它们与4c/A939572相比没有效力优势。芳基3位上存在官能团似乎对具有高效力很重要,因为未取代的类似物显示出显著的活性损失(4k,0.6μM)。酰胺衍生物的微调表明,只有伯酰胺或少量仲酰胺如乙酰胺是有利的,IC50与4c相当。大体积组对活性有害,因为4q在10μM时没有抑制作用。 由于SCD1活性取决于细胞色素b5和细胞色素b5-还原酶,我们建立了一种选择性测定法,以排除被鉴定为SCD1抑制剂的化合物实际上可能抑制辅酶的可能性。当浓度超过10μM时,抑制作用的缺乏支持了我们基于尿素的化学型对SCD的直接抑制,以化合物4c/A939572为代表。 接下来,我们研究了强效抑制剂的选择性。通常可以实现对激酶的优异选择性。4c/A939572也在3H-Dofetilide结合试验中进行了测试,未显示出明显的hERG通道阻断活性(IC50>100μM)[1]。 |
|
| 体内研究 (In Vivo) |
在携带 A498 ccRCC 异种移植物的无胸腺裸鼠 (nu/nu) 中,单独或组合使用 A939572(30 mg/kg,口服)和 Tem 治疗 4 周后测量肿瘤体积 (mm3)。 A939572 和 Tem 单一疗法观察到类似的生长反应;研究结束时,肿瘤体积减少了 20-30%(与安慰剂对照相比);然而,直到治疗的最后一周,数据才达到统计学显着性。当研究结束时,联合治疗组的肿瘤体积减少了 60% 以上(与安慰剂对照组相比),并且在治疗大约一周后观察到显着减少 [2]。
在一项为期5天的ob/ob小鼠疗效研究中,对SCD1抑制剂4b和4c/A939572进行了评估。以16:0/16:1n7或18:0/18:1n9的比率计算的去饱和指数用作活性的体内生物标志物。检查的脂质类别包括胆固醇酯、二酰基甘油、游离脂肪酸、游离胆固醇、总磷脂和三酰基甘油。4b(30mpk-bid)和4c/A939572(10mpk-bid)持续将所有这些脂质类别的去饱和指数降低到瘦水平,甚至更低。图2显示了SCD1抑制剂4b和4c治疗的去饱和指数(18:0/18:1n9)降低效果。在ob/ob小鼠中也观察到甘油三酯去饱和指数降低4c的剂量依赖性(数据未显示)。这些观察结果表明,体外活性与体内去饱和指数之间存在明显的相关性[1]。 |
|
| 酶活实验 |
SCD1体外酶法测定。[1]
通过测量3H(9,10)硬脂酰辅酶A底物去饱和产生的氚水来测定SCD1活性。该检测的一个版本使用ob/ob小鼠肝微粒体作为SCD1酶的来源。另一种使用与人cyt b5/cyt b5R串联表达的重组人SCD1作为SCD1酶的来源。 SCD1活性测定在50℃的96孔板中进行L适用于高通量筛选的总测定反应体积。通常为50L反应体积:5L在10%DMSO和10M硬脂酰辅酶A,0.24M 3H(9,10)硬脂酰辅酶A在35L测定缓冲液(250 mM蔗糖、10 mM Tris pH 7.5、5mM MgCl2、1 mM DTT、罗氏完全EDTA蛋白酶抑制剂混合物、2 mM NADH和50 mM NaF)。然后加入蛋白质浓度在10-20之间的人或小鼠微粒体来引发反应g蛋白质,取决于特定微粒体制剂的比活性。 在30分钟的反应时间后,通过加入终止溶液终止反应,通常为30分钟L 4N HCl。设定的反应体积,通常为65L、 然后从每个孔转移到Millipore多筛板中的50mg甲醇预润湿的Norit A炭中。将板摇动1分钟,静置5分钟,以吸附硬脂酰辅酶A和油酰辅酶A。通过离心将3H-H2O与吸附有木炭的硬脂酰辅酶a和油酰辅酶a分离到收集板中。移除充满木炭的多屏幕板,并向每个孔中加入闪烁液。通常在Microbeta-TriLux放射性计数器中计算放射性。在背景减除后,SCD1活性相对于没有抑制剂的对照反应表示。使用非线性回归分析确定剂量反应曲线拟合和IC50值。 输血和萤光素酶检测[2] 使用Lipofectamine2000用p5xATF6-GL3-UPR荧光素酶报告基因和pRL-CMV肾荧光素酶质粒瞬时转染Caki1和A498细胞。根据制造商的方案,使用Promega的双荧光素酶检测试剂盒在48小时后收获细胞(DMSO vs.A939572NT vs.shSCD780),并使用Veritas光度计测量荧光素酶活性;报告为相对发光。 RNA分离和定量PCR[2] 如前所述进行RNA分离、cDNA制备和QPCR。TaqMan®FAM™染料标记探针,包括POLR2A(Hs00172187_m1-normalization control)、SCD1(Hs01682761_m1)、HSPA5(Hs99999174_m1)、CEBPβ(CEBPB Hs00270923_s1)、GADD45A(Hs0.0169255_m1),DDIT3(Hs01090850_m1);HERPUD1(Hs01124269_m1)和ATF6(Hs00232586_m1)。倍数变化值比较:使用ΔΔCt法比较正常与肿瘤、NT与靶慢病毒、DMSO与A939572处理的样本。 |
|
| 细胞实验 |
将细胞(0.5 或 1×105/孔)一式三份接种在 24 孔板中。使用库尔特粒子计数器对细胞进行计数。将油酸-白蛋白以 5μMol 添加到培养基中。 A939572 库存是在 DMSO 中制备的。进行替西罗莫司给药。软琼脂培养物是通过在 1.5% Seaplaque®GTG® 琼脂糖中以 1:1 的比例稀释 2 倍生长培养基来制备的,在 60mm 培养皿中每板 500 个细胞。 3 周后用吉姆萨 (Giemsa) 对集落进行染色并计数[2]。
|
|
| 动物实验 |
体内分析[2]
将A498细胞以1×10⁶个细胞/只的密度皮下植入无胸腺nu/nu小鼠体内,细胞悬浮于50% Matrigel基质胶中。肿瘤体积在4周治疗前达到约50 mm³。A939572以30 mg/kg的剂量,用草莓味Kool-Aid®(溶于无菌水中,浓度为0.2 g/mL)重悬,每次50 μl。小鼠每日两次通过注射器灌胃给药,每次50 μl。该改良方法有效且对小鼠的应激反应较小。将替西罗莫司溶解于30%乙醇/生理盐水中,每72小时一次,每次50 μl,通过腹腔注射给药,剂量为10 mg/kg。肿瘤体积采用公式 0.5236(长*宽*高) 计算,每 3 天测量一次体重。 |
|
| 药代性质 (ADME/PK) |
总的来说,该系列 SCD1 抑制剂在啮齿动物中具有良好的药代动力学特征,其特点是分布容积适中,在小鼠中具有较高的口服生物利用度(4c/A939572,CLp = 0.4 L/h/kg;Vss = 0.4 L/kg;F = 92%)。
|
|
| 参考文献 | ||
| 其他信息 |
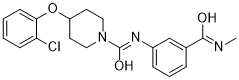
通过分子骨架改造,我们鉴定了一系列结构新颖的硬脂酰辅酶A去饱和酶1 (SCD1) 抑制剂。初步的构效关系 (SAR) 研究发现了高效且口服生物利用度高的哌啶-芳基脲类SCD1抑制剂。4-(2-氯苯氧基)-N-[3-(甲基氨基甲酰基)苯基]哌啶-1-甲酰胺 4c 表现出显著的体内活性,并具有剂量依赖性的去饱和指数降低作用。[1] 目的:我们旨在将硬脂酰辅酶A去饱和酶1 (SCD1) 鉴定为透明细胞肾细胞癌 (ccRCC) 的新型分子靶点,并研究其在体外和体内独立以及与美国食品药品监督管理局 (FDA) 当前批准的治疗方案联合使用时对肿瘤细胞生长和活力的影响。实验设计:检测患者正常组织和ccRCC组织样本以及细胞系中SCD1的表达。本研究利用基因敲低模型和小分子抑制剂A939572靶向抑制SCD1,并采用基因芯片分析方法,分析了SCD1对细胞生长、凋亡和基因表达的影响。此外,我们还评估了使用酪氨酸激酶抑制剂(TKI)舒尼替尼和帕唑帕尼以及mTOR抑制剂替西罗莫司进行SCD1药理学抑制的协同治疗模型。结果显示,SCD1在所有阶段的透明细胞肾细胞癌(ccRCC)中均表达升高。SCD1的基因敲低和药理学抑制均可降低肿瘤细胞增殖,并在体外和体内诱导细胞凋亡。通过基因芯片、定量实时PCR和蛋白质分析A939572处理或SCD1慢病毒敲低样本,我们观察到内质网应激反应信号通路被激活,这为SCD1在ccRCC中的作用机制提供了新的见解。此外,A939572 与替西罗莫司联合应用可协同抑制体外和体内肿瘤生长。结论:SCD1 表达增加支持 ccRCC 细胞的存活,因此我们提出将其作为一种新的分子靶点,可单独或与 mTOR 抑制剂联合用于无法通过手术治疗的患者,例如晚期或转移性疾病患者。[2]
|
| 分子式 |
C20H22CLN3O3
|
|---|---|
| 分子量 |
387.8600
|
| 精确质量 |
387.135
|
| 元素分析 |
C, 61.93; H, 5.72; Cl, 9.14; N, 10.83; O, 12.37
|
| CAS号 |
1032229-33-6
|
| PubChem CID |
24905400
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.302 g/cm3
|
| 沸点 |
633.5ºC at 760 mmHg
|
| 闪点 |
336.9ºC
|
| LogP |
4.176
|
| tPSA |
70.67
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
511
|
| 定义原子立体中心数目 |
0
|
| SMILES |
ClC1=C([H])C([H])=C([H])C([H])=C1OC1([H])C([H])([H])C([H])([H])N(C(N([H])C2=C([H])C([H])=C([H])C(C(N([H])C([H])([H])[H])=O)=C2[H])=O)C([H])([H])C1([H])[H]
|
| InChi Key |
DPYTYQFYDLYWHZ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C20H22ClN3O3/c1-22-19(25)14-5-4-6-15(13-14)23-20(26)24-11-9-16(10-12-24)27-18-8-3-2-7-17(18)21/h2-8,13,16H,9-12H2,1H3,(H,22,25)(H,23,26)
|
| 化学名 |
4-(2-chlorophenoxy)-N-[3-(methylcarbamoyl)phenyl]piperidine-1-carboxamide
|
| 别名 |
A939572; A-939572; 1032229-33-6; A939,572; 4-(2-chlorophenoxy)-N-[3-(methylcarbamoyl)phenyl]piperidine-1-carboxamide; 4-(2-chlorophenoxy)-N-(3-(methylcarbamoyl)phenyl)piperidine-1-carboxamide; 4-(2-CHLOROPHENOXY)-N-[3-[(METHYLAMINO)CARBONYL]PHENYL]-1-PIPERIDINECARBOXAMIDE; 1-Piperidinecarboxamide,4-(2-chlorophenoxy)-N-[3-[(methylamino)carbonyl]phenyl]-; CHEMBL469169; A 939,572; A 939572; SCD1 Inhibitor; Stearoyl-CoA Desaturase 1 Inhibitor;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~257.82 mM)
H2O : < 0.1 mg/mL |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.45 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.45 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.45 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5782 mL | 12.8912 mL | 25.7825 mL | |
| 5 mM | 0.5157 mL | 2.5782 mL | 5.1565 mL | |
| 10 mM | 0.2578 mL | 1.2891 mL | 2.5782 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
