| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
CX3CR1 ( Ki = 3.9 nM ); 125I-IL-8-CXCR2 ( Ki = 2800 nM )
C-X-C motif chemokine receptor 1 (CX3CR1) (Ki = 0.8 nM for human CX3CR1; IC₅₀ = 1.1 nM for inhibiting CX3CL1 binding to human CX3CR1; IC₅₀ = 2.3 nM for inhibiting CX3CR1-mediated calcium mobilization); >1000-fold selectivity over CXCR1, CXCR2, CXCR3, CXCR4, CCR1, CCR2, CCR5, CCR7 (Ki > 1000 nM for all) [1][3] |
|
|---|---|---|
| 体外研究 (In Vitro) |
体外活性:AZD8797是人CX3CR1受体的选择性小分子和变构非竞争性调节剂;它拮抗 CX3CR1 和 CXCR2,Ki 分别为 3.9 和 2800 nM。在流动粘附测定中,AZD8797 拮抗人全血 (hWB) 和 B 淋巴细胞细胞系中的天然配体 fractalkine (CX3CL1),IC50 值分别为 300 和 6 nM。 AZD8797 还在 [(35)S]GTPγS(鸟苷 5-[γ-硫代]三磷酸)积累测定中阻止 G 蛋白激活。相反,动态质量再分配(DMR)实验揭示了弱的 Gαi 依赖性 AZD8797 激动作用。此外,在 β-抑制蛋白招募试验中,AZD8797 在亚微摩尔浓度下正向调节 CX3CL1 反应。在平衡饱和结合实验中,AZD8797 降低了 (125)I-CX3CL1 的最大结合而不影响 Kd。确定 AZD8797 的 kon 和 koff 的动力学实验表明,这不是不可逆或不可克服的结合的假象,因此是真正的非竞争性机制。最后,我们发现 AZD8797 和 GTPγS 都以类似的方式增加 CX3CL1 与 CX3CR1 解离的速率,表明 AZD8797 和 CX3CR1 结合的 G 蛋白之间存在联系。总的来说,这些数据表明 AZD8797 是 CX3CL1 的非竞争性变构调节剂,结合 CX3CR1 并以偏向方式影响 G 蛋白信号传导和 β-抑制蛋白招募。激酶测定:将 CHO-hCX3CR1 膜与不同浓度的 AZD8797 一起在 MicroWell 96 孔板中的 50 mM HEPES、100 mM NaCl、5 mM MgCl2、10 μM GDP 和 0.01% 明胶中孵育。然后添加 0.56 μCi/mL [35S]GTPγS 和 CX3CL1 的 EC80。将板在 30°C 下孵育 1 小时,然后通过真空过滤将未结合的 [35S]GTPγS 从结合中分离到 Printed Filtermat B。通过在 DMSO 中逐步稀释以达到 1 的最终 DMSO 浓度,可实现不同的 AZD8797 浓度。添加测定缓冲液后所有孔中的百分比,无论 AZD8797 浓度如何。细胞测定:在流动粘附测定中,AZD8797 拮抗人全血 (hWB) 和 B 淋巴细胞细胞系中的天然配体 fractalkine (CX3CL1),IC50 值分别为 300 和 6 nM。 AZD8797 还可防止 [35S]GTPγS 积累测定中的 G 蛋白激活。在 β-抑制蛋白招募试验中,AZD8797 在亚微摩尔浓度下正向调节 CX3CL1 反应。在平衡饱和结合实验中,AZD8797 降低了 125I-CX3CL1 的最大结合而不影响 Kd。 AZD8797 以高亲和力选择性地结合人和大鼠 CX3CR1(hCX3CR1 的 Ki,4 nM;rCX3CR1 的 Ki,分别为 7 nM)。平衡解离常数 KB 表明 AZD8797 是人类 CX3CR1 的非常有效的抑制剂 (10 nM)。大鼠 CX3CR1 的效力低三倍 (29 nM),小鼠 CX3CR1 的效力进一步降低 (54 nM)。
CX3CR1结合及变构调节:AZD8797是人CX3CR1的强效、高选择性变构非竞争性调节剂,对人CX3CR1的结合亲和力Ki=0.8 nM。它不与内源性配体CX3CL1竞争正构位点,而是通过诱导受体构象变化抑制CX3CL1介导的受体激活;以剂量依赖性方式阻断CX3CL1与人CX3CR1的结合,IC₅₀=1.1 nM[1][3] - CX3CR1功能抑制:在稳定表达人CX3CR1的CHO细胞中,AZD8797抑制CX3CL1诱导的钙流,IC₅₀=2.3 nM;10 nM浓度下抑制CX3CL1介导的ERK1/2磷酸化78%,减少细胞内cAMP积累65%,且不影响基础信号传导。在人外周血单核细胞和THP-1细胞中,阻断CX3CL1诱导的趋化,IC₅₀值分别为3.5 nM和4.2 nM[1][3] - 种属交叉反应性:该化合物对大鼠CX3CR1的Ki=4.6 nM,对小鼠CX3CR1的Ki=5.8 nM,与啮齿类同源受体表现出中等交叉反应性[2][3] - 代谢稳定性:人肝微粒体中,AZD8797表现出良好的代谢稳定性,半衰期(t₁/₂)为85分钟,内在清除率(CLint)为17 μL/min/mg蛋白[3] |
|
| 体内研究 (In Vivo) |
AZD8797 对患有髓磷脂少突胶质细胞糖蛋白诱导的 EAE 的 Dark Agouti 大鼠进行治疗,可减少麻痹、中枢神经系统病理学和复发率。该化合物在发病前以及急性期后开始治疗时均有效。
大鼠实验性自身免疫性脑脊髓炎(EAE)模型(慢性复发性多发性硬化症模型):免疫后第7天至第21天,口服给予AZD8797(3、10、30 mg/kg,每日一次),剂量依赖性减轻疾病严重程度。30 mg/kg剂量下,平均最大临床评分为1.2(溶媒组为3.8),复发频率降低68%。脊髓组织学分析显示,炎症细胞浸润减少(30 mg/kg剂量下CD4+ T细胞和巨噬细胞分别减少62%和58%),脱髓鞘减轻70%,轴索损伤减少65%;流式细胞术证实CX3CR1+免疫细胞向中枢神经系统(CNS)的募集减少[2] - 大鼠药效学研究:口服AZD8797(10 mg/kg)后,脑内药物浓度可持续维持(给药后4小时为0.7 μg/g),在LPS激发的大鼠中抑制CX3CR1介导的单核细胞向CNS浸润55%[3] |
|
| 酶活实验 |
在 MicroWell 96 孔板中,将 CHO-hCX3CR1 膜在 50 mM HEPES、100 mM NaCl、5 mM MgCl2、10 μM GDP 和 0.01% 明胶以及不同浓度的 AZD8797 中孵育。接下来,添加CX3CL1的EC80和0.56μCi/mL[35S]GTPnS。 30°C 孵育一小时后,将板真空过滤至 Printed Filtermat B,以分离结合和未结合的 [35S]GTPγS。无论 AZD8797 浓度如何,各种 AZD8797 浓度都是通过在 DMSO 中逐步稀释获得的,这导致添加测定缓冲液后所有孔中的最终 DMSO 浓度为 1%。
CX3CR1放射性配体结合实验:制备表达人CX3CR1的HEK293细胞细胞膜,悬浮于结合缓冲液(三羟甲基氨基甲烷-盐酸、氯化镁、0.1%牛血清白蛋白)中。将系列稀释(0.001–1000 nM)的AZD8797与细胞膜及氚标记CX3CL1混合,25°C孵育120分钟后,通过预湿润的玻璃纤维滤膜过滤分离结合态与游离态配体。滤膜用冷结合缓冲液洗涤后,液体闪烁计数器测量放射性强度,通过置换曲线的非线性回归分析计算Ki/IC₅₀值[1][3] - 变构结合表面等离子体共振(SPR)实验:将重组人CX3CR1固定于CM5传感芯片,AZD8797(0.1–100 nM)以30 μL/min流速注入运行缓冲液(HBS-EP+)中。通过1:1结合模型拟合传感图确定结合动力学参数(kon、koff、KD);将CX3CL1(10 nM)与AZD8797共注射,证实非竞争性变构相互作用——CX3CL1结合亲和力无变化,但受体激活被抑制[1] - 受体选择性实验:按上述方法制备表达人CXCR1、CXCR2、CXCR3、CXCR4、CCR1、CCR2、CCR5或CCR7的细胞膜,AZD8797测试浓度最高达10 μM,测定结合亲和力(Ki)以评估对非靶标趋化因子受体的选择性[3] |
|
| 细胞实验 |
在人全血 (hWB) 和 B 淋巴细胞细胞系中,AZD8797 在流动粘附测定中抑制天然配体 fractalkine (CX3CL1),IC50 值分别为 300 和 6 nM。此外,AZD8797 在 [35S]GTPγS 积累测定中抑制 G 蛋白激活。在 β-arrestin 募集测定中,亚微摩尔浓度的 AZD8797 正向调节 CX3CL1 反应。 AZD8797 在平衡饱和结合实验中降低 125I-CX3CL1 的最大结合,但对 Kd 没有影响。 AZD8797 在人类和大鼠中与 CX3CR1 结合时表现出高亲和力和选择性(hCX3CR1 的 Ki 为 4 nM,rCX3CR1 的 Ki 为 7 nM)。平衡解离常数 KB 证明了 AZD8797 是一种非常强的人类 CX3CR1 抑制剂 (10 nM)。大鼠 CX3CR1 的效力为 29 nM,比小鼠 CX3CR1 低三倍,而小鼠 CX3CR1 的效力更小,为 54 nM。
CX3CR1介导钙流检测实验:表达CX3CR1的CHO细胞用钙敏感荧光染料(Fura-2 AM)负载45分钟(37°C)。AZD8797(0.01–100 nM)与细胞预孵育20分钟后,加入CX3CL1(10 nM)刺激。使用酶标仪实时测量荧光强度(激发光340/380 nm,发射光510 nm),通过剂量-反应曲线推导IC₅₀值[1] - 人单核细胞/THP-1细胞趋化实验:人外周血单核细胞或THP-1细胞悬浮于RPMI 1640培养基。AZD8797(0.1–100 nM)与细胞混合后加入Transwell插入物(5 μm孔径)上室,下室加入CX3CL1(10 nM),37°C、5% CO₂孵育3小时。血细胞计数板计数下室中的迁移细胞,计算相对于溶媒对照组的抑制率[3] - CX3CR1信号通路实验:将表达CX3CR1的HEK293细胞以2×10⁶个细胞/孔接种到6孔板,孵育过夜。细胞用AZD8797(10 nM)预处理30分钟后,加入CX3CL1(10 nM)刺激15分钟。用含蛋白酶/磷酸酶抑制剂的RIPA缓冲液裂解细胞,蛋白质印迹法检测磷酸化ERK1/2、总ERK1/2及GAPDH(内参)的表达[1] |
|
| 动物实验 |
|
|
| 药代性质 (ADME/PK) |
在大鼠中:口服给药(10 mg/kg)导致血浆峰浓度(Cₘₐₓ)为 1.8 μg/mL,达到 Cₘₐₓ 的时间(Tₘₐₓ)为 1.5 小时,末端半衰期(t₁/₂)为 6.2 小时,分布容积(Vd)为 3.6 L/kg,口服生物利用度为 58%。静脉注射(5 mg/kg)显示清除率(CL)为 0.47 L/h/kg [3]
- 中枢神经系统渗透:在大鼠中,口服给药(10 mg/kg)2 小时后,AZD8797 达到脑浓度 0.9 μg/g,脑血浆比为 0.7,证实了其有效穿透血脑屏障 [3] - 组织分布:大鼠口服给药(10 mg/kg)2 小时后,显示药物优先分布于肝脏(组织血浆比 = 2.8)、脾脏(2.5)、肺(2.3)、肾脏(2.0)和脊髓(1.2)[3] - 体外代谢:在大鼠肝微粒体中,AZD8797 的代谢半衰期为 92 分钟;主要代谢途径包括羟基化和硫酸化,未检测到有毒代谢物[3] |
|
| 毒性/毒理 (Toxicokinetics/TK) |
血浆蛋白结合率:AZD8797在人血浆中的血浆蛋白结合率为93%,在大鼠血浆中的血浆蛋白结合率为91%(通过超滤法测定)[3]
- 急性毒性:在大鼠和小鼠中,口服LD₅₀ >200 mg/kg。在为期7天的急性研究中,剂量高达100 mg/kg时未观察到明显的毒性(体重减轻、惊厥、死亡)[3] - 亚慢性毒性:在为期28天的大鼠重复口服给药研究(10、30、100 mg/kg/天)中,AZD8797未引起体重、血液学参数或肝肾功能的显著变化。主要器官(肝脏、肾脏、心脏、肺脏、脑)未发现组织病理学异常[3] - 药物相互作用:体外研究表明,浓度高达 10 μM 时,未观察到对细胞色素 P450 酶(CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4)的抑制作用[3] |
|
| 参考文献 |
|
|
| 其他信息 |
AZD8797是一种强效、选择性、口服生物利用度高的CX3CR1变构非竞争性拮抗剂,属于取代的7-氨基-5-硫代噻唑并[4,5-d]嘧啶类化合物[3]。
其核心作用机制是与CX3CR1上的变构位点(不同于CX3CL1的正构位点)结合,诱导受体构象变化,从而阻断下游G蛋白介导的信号传导(钙动员、ERK1/2激活),而不影响配体结合[1]。 临床前数据支持其在治疗中枢神经系统(CNS)脱髓鞘疾病(例如多发性硬化症)和其他CX3CR1介导的炎症性疾病方面的潜在应用价值,其作用机制是通过抑制促炎性CX3CR1+免疫细胞(单核细胞、T细胞)向CNS的浸润,并减少神经炎症、脱髓鞘和轴突损伤[2] - 该化合物能有效穿透血脑屏障,是治疗中枢神经系统疾病的关键优势,因为大多数CX3CR1拮抗剂缺乏足够的脑部进入能力[2][3] - 对CX3CR1的高选择性最大限度地减少了对其他趋化因子受体的脱靶效应,从而降低了全身免疫功能障碍的风险[1][3] |
| 分子式 |
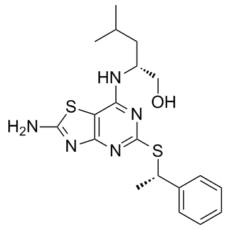
C19H25N5OS2
|
|
|---|---|---|
| 分子量 |
403.56
|
|
| 精确质量 |
403.15
|
|
| 元素分析 |
C, 56.55; H, 6.24; N, 17.35; O, 3.96; S, 15.89
|
|
| CAS号 |
911715-90-7
|
|
| 相关CAS号 |
|
|
| PubChem CID |
11965767
|
|
| 外观&性状 |
Light yellow to yellow solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
651.1±65.0 °C at 760 mmHg
|
|
| 闪点 |
347.6±34.3 °C
|
|
| 蒸汽压 |
0.0±2.0 mmHg at 25°C
|
|
| 折射率 |
1.669
|
|
| LogP |
4.6
|
|
| tPSA |
151
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
8
|
|
| 可旋转键数目(RBC) |
8
|
|
| 重原子数目 |
27
|
|
| 分子复杂度/Complexity |
452
|
|
| 定义原子立体中心数目 |
2
|
|
| SMILES |
N(C1N=C(S[C@H](C2C=CC=CC=2)C)N=C2N=C(SC=12)N)[C@@H](CO)CC(C)C
|
|
| InChi Key |
ZMQSLMZOWVGBSM-GXTWGEPZSA-N
|
|
| InChi Code |
InChI=1S/C19H25N5OS2/c1-11(2)9-14(10-25)21-16-15-17(22-18(20)27-15)24-19(23-16)26-12(3)13-7-5-4-6-8-13/h4-8,11-12,14,25H,9-10H2,1-3H3,(H3,20,21,22,23,24)/t12-,14+/m0/s1
|
|
| 化学名 |
(2R)-2-[[2-amino-5-[(1S)-1-phenylethyl]sulfanyl-[1,3]thiazolo[4,5-d]pyrimidin-7-yl]amino]-4-methylpentan-1-ol
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.19 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.19 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.19 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 5 mg/mL (12.39 mM) in 20% HP-β-CD in Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4779 mL | 12.3897 mL | 24.7795 mL | |
| 5 mM | 0.4956 mL | 2.4779 mL | 4.9559 mL | |
| 10 mM | 0.2478 mL | 1.2390 mL | 2.4779 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 The CX3CR1 inhibitor AZD8797 blocks MOG-EAE in DA rats.Proc Natl Acad Sci U S A.2014 Apr 8;111(14):5409-14. |
|---|
 AZD8797 treatment in EAE reduces TSPO binding, a clinically relevant marker for microglia activation.Proc Natl Acad Sci U S A.2014 Apr 8;111(14):5409-14. |
 AZD8797 treatment in rats with established EAE inhibits development of relapses and all histopathological manifestations of the disease.Proc Natl Acad Sci U S A.2014 Apr 8;111(14):5409-14. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
