| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
BAY-1436032 is a brand-new pan-mutant inhibitor of isocitrate dehydrogenase 1 (IDH1). In mouse hematopoietic cells expressing IDH1R132H or IDH1R132C, BAY-1436032 reduces intracellular (R)-2-hydroxyglutarate (R-2HG) synthesis with IC50 values of 60 and 45 nM, respectively. Even at dosages up to 10 μM, BAY-1436032 does not lower R-2HG levels in mouse hematopoietic cells expressing IDH2R140Q and IDH2R172K. At doses as high as 100 μM, BAY-1436032 did not impede the colony expansion of patient-derived IDH1 wild-type AML cells, but it did at 50% when it came to that growth. BAY-1436032 significantly enhanced myelomonocytic differentiation of myeloid progenitor cells in morphological assessment [1].
|
|---|---|
| 体外研究 (In Vitro) |
BAY-1436032是一种全新的异柠檬酸脱氢酶1(IDH1)全突变抑制剂。在表达 IDH1R132H 或 IDH1R132C 的小鼠造血细胞中,BAY-1436032 减少细胞内 (R)-2-羟基戊二酸 (R-2HG) 的合成,IC50 值分别为 60 和 45 nM。即使剂量高达 10 μM,BAY-1436032 也不会降低表达 IDH2R140Q 和 IDH2R172K 的小鼠造血细胞中的 R-2HG 水平。在高达 100 μM 的剂量下,BAY-1436032 不会阻碍患者来源的 IDH1 野生型 AML 细胞的集落扩张,但在增长时会阻碍 50%。 BAY-1436032 在形态学评估中显着增强了骨髓祖细胞的骨髓单核细胞分化 [1]。
BAY-1436032 选择性抑制表达 IDH1R132H(IC₅₀ 60 nM)或 IDH1R132C(IC₅₀ 45 nM)的永生化小鼠造血细胞内的 R-2HG 产生。[1] BAY-1436032 能有效抑制携带各种 IDH1R132 突变的人原代 AML 细胞中 R-2HG 的产生(IC₅₀ 在 3 到 16 nM 之间),但在浓度高达 1 μM 时对 IDH2 突变细胞几乎没有影响。[1] BAY-1436032 在甲基纤维素克隆形成实验中抑制人原代 IDH1 突变 AML 细胞的集落形成,在 0.1 μM 浓度下抑制约 50%。即使浓度高达 100 μM,也不抑制 IDH1 野生型 AML 细胞的集落生长。[1] BAY-1436032 在原代 IDH1 突变 AML 细胞的体外培养中诱导髓系分化,表现为表面分化标志物 CD14 和 CD15 的上调,以及形态学上显示单核细胞/粒细胞成熟的改变。[1] BAY-1436032 处理(500 nM,14 天)降低了原代 IDH1R132C 和 IDH1R132H 突变 AML 细胞中组蛋白 H3 在 H3K4、H3K9、H3K27 和 H3K36 位点的全局三甲基化水平,但对 IDH1 野生型细胞无此作用。[1] 使用 Illumina Infinium HumanMethylation450 平台进行的 DNA 甲基化分析表明,BAY-1436032 处理(500 nM,14 天)改变了特定的甲基化模式,包括降低 IDH1 突变 AML 细胞中 PLU1 启动子的甲基化和增加 E2F1 启动子的甲基化。[1] |
| 体内研究 (In Vivo) |
在 150 mg/kg 剂量下,每天一次长期口服 BAY-1436032 治疗显示几乎完全抑制 (R)-2-羟基戊二酸 (R-2HG) 合成。接受载体治疗的小鼠的白细胞计数稳步上升;在给予 45 mg/kg BAY-1436032 的小鼠中,增加速度减慢;在 150 mg/kg 组中,数字没有变化。第 60 天,媒介物组和 45 mg/kg 组的血红蛋白水平略低于 150 mg/kg 组,而用 BAY-1436032 以 45 mg/kg 组和媒介物组治疗的小鼠的血红蛋白水平略低于 150 mg/kg 组。 150毫克组的动物。第 60 天,该队列的血小板计数/公斤显着减少。虽然用媒介物治疗的动物死亡,但所有给予 150 mg/kg BAY-1436032 的小鼠均存活,并且外周血中的 hCD45+ 细胞负荷最低,直至治疗开始后第 150 天观察结束(P<0.001)。位的使用寿命为 91 天。 45 mg/kg 剂量的 BAY-1436032 在小鼠体内产生中等表达的 CD14/CD15 [1]。
在 IDH1R132C 突变 AML 的患者来源异种移植(PDX1)小鼠模型中,移植后 17 天开始,每日一次口服 150 mg/kg 的 BAY-1436032,持续 150 天,导致血清 R-2HG 水平持续抑制,外周血中人 CD45⁺ (hCD45⁺) 白血病母细胞显著减少,并延长了生存期(所有小鼠存活至第 150 天,而溶剂组中位生存期为 91 天)。[1] 在第二个独立的、伴有 MLL-PTD 共突变的 IDH1R132C 突变 AML PDX 模型(PDX2)中,移植后 90 天开始,每日一次口服 150 mg/kg BAY-1436032 治疗 100 天,抑制了血清 R-2HG,降低了外周血 hCD45⁺ 母细胞计数,并显著延长了生存期(10 只小鼠中有 6 只存活至治疗停止的第 100 天,中位生存期未达到,而溶剂组为 62 天)。[1] BAY-1436032 在体内诱导白血病细胞髓系分化,表现为经治疗的 PDX 小鼠外周血中表达 CD14 和 CD15 的 hCD45⁺ 细胞比例增加,以及 CD34⁺ 祖细胞比例减少。[1] BAY-1436032 在体内耗竭了白血病干细胞(LSCs)。有限稀释移植实验显示,与溶剂处理的小鼠相比,用 BAY-1436032(150 mg/kg)治疗的小鼠骨髓细胞中 LSC 频率降低了约 100 倍。[1] 对来自治疗后的 PDX 小鼠的 hCD45⁺ 细胞进行的基因表达谱分析显示,干性相关基因(如 SPARC、CD69、DNMT3B)下调,而髓系分化和免疫反应相关基因上调。细胞周期分析显示,BAY-1436032 治疗后,处于 G₀/G₁ 期的 hCD45⁺ 细胞增加,S 期细胞减少。[1] |
| 细胞实验 |
为了评估 R-2HG 产生的抑制,用逆转录病毒工程化表达突变 IDH1 或 IDH2 的 HoxA9 永生化小鼠骨髓细胞用 BAY-1436032 或 DMSO 处理 8 天。收获细胞,通过质谱法测量细胞裂解液中 R-2HG 与 S-2-羟基戊二酸(S-2HG)的比值。[1]
从患者新鲜分离的原代人 AML 细胞用 BAY-1436032 或 DMSO 处理 24 小时。测量细胞裂解液中 R-2HG 与 S-2HG 的比值以确定 IC₅₀ 值。[1] 对于克隆形成祖细胞实验,将人原代 AML 单核细胞接种在补充了细胞因子以及 BAY-1436032 或溶剂的甲基纤维素培养基中。10-14 天后在显微镜下计数集落。[1] 为了评估分化,将原代 IDH1 突变 AML 细胞在含有 BAY-1436032 或 DMSO 的悬浮培养基中培养数天。然后通过流式细胞术分析 CD14 和 CD15 的表达,并在染色后通过细胞涂片进行形态学评估。[1] 对于组蛋白甲基化分析,原代 AML 细胞用 500 nM BAY-1436032 或 DMSO 处理 14 天。提取组蛋白,通过 Western blotting 分析 H3K4me3、H3K9me3、H3K27me3 和 H3K36me3 的全局水平,并定量条带强度相对于总组蛋白 H3 的比值。[1] |
| 动物实验 |
在药代动力学和药效学研究中,小鼠分别以 45、90 或 150 mg/kg 的剂量经灌胃给予 BAY-1436032。在不同时间点采集血液样本,以测定血浆药物浓度和血清 R-2HG 水平。[1]
在 PDX 模型中开展疗效研究时,将原代人 IDH1 突变型 AML 细胞静脉移植到 NOD-scid IL2Rγnull (NSG) 小鼠体内。一旦移植成功(以外周血中 hCD45⁺ 细胞数量为指标),即开始治疗。[1] 在 PDX1 模型中,治疗于移植后 17 天开始。小鼠(每组 n=10)每天一次经灌胃给予载体、45 mg/kg 或 150 mg/kg 的 BAY-1436032,持续 150 天。 [1] 在PDX2模型中,为模拟较高的疾病负荷,治疗于移植后90天开始。小鼠(每组n=10)每日一次灌胃给予载体或150 mg/kg的BAY-1436032,持续100天,之后停止治疗并监测小鼠的复发情况。[1] 监测小鼠的存活率、体重和外周血细胞计数。定期通过流式细胞术分析外周血中hCD45⁺细胞和分化标志物(CD14、CD15、CD34、CD33)。采用质谱法或酶法测定血清R-2HG水平。[1] 为了进行极限稀释试验以评估LSC频率,从接受载体或BAY-1436032(150 mg/kg)治疗4周的PDX小鼠中收集骨髓细胞。将系列稀释的细胞移植到次级NSG受体中,并评估移植情况,使用泊松统计方法计算LSC频率。[1] |
| 药代性质 (ADME/PK) |
在小鼠体内,BAY-1436032的血浆暴露量在45至150 mg/kg剂量范围内几乎呈剂量线性关系。[1]
给药后24小时内,游离血浆浓度覆盖了体外R-2HG抑制的IC₅₀值。[1] 口服BAY-1436032可导致血清R-2HG水平迅速下降,最早可在给药后3小时检测到。[1] 长期每日一次口服150 mg/kg BAY-1436032可使PDX小鼠体内R-2HG的生成几乎完全受到抑制。[1] |
| 参考文献 | |
| 其他信息 |
泛突变型IDH1抑制剂Bay-1436032是一种口服的泛抑制剂,可抑制代谢酶异柠檬酸脱氢酶1型(IDH1;IDH-1;IDH1 [NADP+] 可溶性)的突变形式,包括精氨酸132位点突变的IDH1(IDH1(R132)),具有潜在的抗肿瘤活性。给药后,泛突变型IDH-1抑制剂BAY-1436032特异性抑制IDH1突变体的活性,从而阻止α-酮戊二酸(α-KG)生成致癌代谢物2-羟基戊二酸(2HG)。这阻断了2HG介导的信号传导,导致表达IDH1突变体的肿瘤细胞分化增强和增殖抑制。 IDH1 突变,包括 IDH1(R132) 突变,在某些恶性肿瘤中高表达;它们通过阻断细胞分化和催化 2-羟基戊二酸 (2HG) 的生成来启动和驱动癌症生长。
BAY-1436032 是一种新型的口服生物利用度高的泛突变型 IDH1 抑制剂,旨在靶向所有在癌症中发现的主要 IDH1R132 突变。[1] 其作用机制包括抑制突变型 IDH1 的新功能活性,从而减少致癌代谢物 R-2HG 的产生。这导致组蛋白和 DNA 高甲基化逆转、分化阻滞解除、增殖抑制和白血病干细胞耗竭。 [1] 该研究表明,BAY-1436032在体内对人类IDH1突变型急性髓系白血病具有高效性,可清除原始细胞,促进细胞分化,并在临床前PDX模型中延长生存期。[1] 基于这项临床前研究,在本文发表时,一项针对IDH1R132突变型AML患者的I期临床试验正在启动。[1] |
| 分子式 |
C26H30F3N3O3
|
|---|---|
| 分子量 |
489.529917240143
|
| 精确质量 |
489.22
|
| 元素分析 |
C, 63.79; H, 6.18; F, 11.64; N, 8.58; O, 9.80
|
| CAS号 |
1803274-65-8
|
| PubChem CID |
118310260
|
| 外观&性状 |
White to gray solid powder
|
| LogP |
7.2
|
| tPSA |
76.4
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
35
|
| 分子复杂度/Complexity |
726
|
| 定义原子立体中心数目 |
2
|
| SMILES |
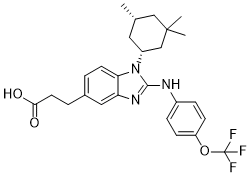
C[C@H]1C[C@H](CC(C1)(C)C)N2C3=C(C=C(C=C3)CCC(=O)O)N=C2NC4=CC=C(C=C4)OC(F)(F)F
|
| InChi Key |
RNMAUIMMNAHKQR-QFBILLFUSA-N
|
| InChi Code |
InChI=1S/C26H30F3N3O3/c1-16-12-19(15-25(2,3)14-16)32-22-10-4-17(5-11-23(33)34)13-21(22)31-24(32)30-18-6-8-20(9-7-18)35-26(27,28)29/h4,6-10,13,16,19H,5,11-12,14-15H2,1-3H3,(H,30,31)(H,33,34)/t16-,19+/m0/s1
|
| 化学名 |
3-(2-((4-(trifluoromethoxy)phenyl)amino)-1-((1R,5R)-3,3,5-trimethylcyclohexyl)-1H-benzo[d]imidazol-5-yl)propanoic acid
|
| 别名 |
BAY-1436032; BAY 1436032; BAY1436032.
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~125 mg/mL (~255.35 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.11 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (5.11 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.11 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0428 mL | 10.2139 mL | 20.4278 mL | |
| 5 mM | 0.4086 mL | 2.0428 mL | 4.0856 mL | |
| 10 mM | 0.2043 mL | 1.0214 mL | 2.0428 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
BAY1436032 inhibits proliferation and induces myeloid differentiation in patient-derived IDH1 mutant AML cellsex vivo.Leukemia. 2017 Oct;31(10):2020-2028. |
BAY1436032 induces myeloid differentiation of IDH1R132C mutant AML cellsin vivo.Leukemia. 2017 Oct;31(10):2020-2028. |
BAY1436032 impacts on histone methylation in patient-derived IDH1R132C and IDH1R132H mutant AML cellsex vivo. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
