| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
CCR5 ( IC50 = 0.29 nM ); CCR2 ( IC50 = 5.9 nM ); R5 HIV-1 ( IC50 = 0.024-0.08 nM ); R5 HIV-2 ( IC50 = 0.03-0.98 nM )
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:Cenicriviroc(以前称为 TAK-652 或 TBR-652)是一种新型口服生物活性 CCR2/CCR5 双重拮抗剂,它还能抑制 HIV-1 和 HIV-2,并具有潜在的治疗艾滋病毒感染。 TAK-652 在纳摩尔浓度下抑制 RANTES(受激活、正常 T 细胞表达和分泌调节)、巨噬细胞炎症蛋白 1α (MIP-1α) 和 MIP-1β 与 CCR5 表达细胞的结合。 TAK-652 还可以抑制单核细胞趋化蛋白 1 (MCP-1) 与表达 CCR2b 的细胞的结合。然而,其对配体与其他趋化因子受体结合的抑制作用有限。 TAK-652 对使用 CCR5(R5)的 HIV-1 有活性,但对使用 CXCR4(X4)的 HIV-1 完全没有活性。激酶测定:Cenicriviroc 可防止人类免疫缺陷病毒 1 型 (HIV-1) 进入细胞,对于测试的 4 个 R5 HIV-2 临床分离株,有效浓度 50% EC50 为 0.03、0.33、0.45 和 0.98 nM。双向性和 X4 向性 HIV-2 毒株对西尼昔韦罗具有耐药性,EC50 > 1000 nM,MPI 分别为 33% 和 4%。细胞测定:AK-652 在纳摩尔浓度下抑制 RANTES(受激活、正常 T 细胞表达和分泌调节)、巨噬细胞炎症蛋白 1α (MIP-1α) 和 MIP-1β 与 CCR5 表达细胞的结合。 TAK-652 还可以抑制单核细胞趋化蛋白 1 (MCP-1) 与表达 CCR2b 的细胞的结合。
|
| 体内研究 (In Vivo) |
Cenicriviroc (≥20 mg/kg/day) 显着减少体内单核细胞/巨噬细胞的募集。在这些剂量下,cenicriviroc 显示出抗纤维化作用,在三种纤维化动物模型中显着减少胶原蛋白沉积以及 1 型胶原蛋白和 mRNA 表达。在 NASH 模型中,cenicriviroc 显着降低非酒精性脂肪肝疾病活动评分。 Cenicriviroc 治疗对身体或肝/肾重量没有显着影响。
Cenicriviroc(CVC)在剂量≥20mg/kg/天时,显著降低了体内单核细胞/巨噬细胞的募集(p<0.05)。在这些剂量下,CVC显示出抗纤维化作用,在三种纤维化动物模型中,胶原沉积(p<0.05)和1型胶原蛋白和mRNA表达显著降低。在NASH模型中,CVC显著降低了非酒精性脂肪肝活动评分(与对照组相比,p<0.05)。CVC治疗对体重或肝肾重量没有显著影响。 结论:CVC在一系列动物纤维化模型中显示出强大的抗炎和抗纤维化活性,支持人类对纤维化疾病的测试。需要进一步的实验研究来阐明CVC抗纤维化作用的潜在机制。一项针对NASH和肝纤维化成年人的2b期研究已全部纳入(CENTAUR研究652-2-203;NCT02217475)。[1] 受试者按每剂量水平4:1的比例随机分配到Cenicriviroc/TBR-652(25、50、75、100或150 mg)或安慰剂组,每天服用一次,持续10天。在第40天测量HIV-1 RNA和CD4细胞计数与基线的变化,在第10天测量单核细胞趋化蛋白-1(MCP-1)、高敏C反应蛋白(hs-CRP)和IL-6的变化。药代动力学数据采用非房室统计进行分析。记录实验室和临床不良事件(AE)和心电图变化。 结果:25、50、75和150 mg剂量的HIV-1 RNA值的最大中值降低分别为-0.7、-1.6、-1.8和-1.7 log10拷贝/毫升。所有变化都很显著。达到最低点的中位时间为10-11天。抑制一直持续到治疗后。50mg和150mg剂量组的平均MCP-1在第10天显著增加。对CD4细胞计数、hs-CRP和IL-6水平的影响可以忽略不计。TBR-652总体上是安全的,耐受性良好,没有因不良事件而停药。 结论:TBR-652在所有剂量下均能显著降低HIV-1 RNA。MCP-1水平的显著升高表明CCR2的阻断作用很强。TBR-652通常耐受良好,没有剂量限制性不良事件。药效学表明,TBR-652作为一种未经强化的每日一次口服CCR5拮抗剂,具有潜在的重要CCR2介导的抗炎作用,值得进一步研究。[4] |
| 酶活实验 |
对于测试的四种 R5 HIV-2 临床分离株,Cenicriviroc 以 50% EC50 为 0.03、0.33、0.45 和 0.98 nM 的有效浓度阻止 HIV-1 进入细胞。双向性和 X4 向性 HIV-2 毒株中的 Cenicriviroc 耐药性对于 EC50 为 >1000 nM,对于 MPI 分别为 33% 和 4%。
趋化因子结合试验。[5] 之前已经描述了测试化合物对趋化因子结合抑制的测定程序。简而言之,表达CCR5的CHO细胞与不同浓度的Cenicriviroc (CVC)/TAK-652在结合缓冲液(含20 mM HEPES和0.5%牛血清白蛋白的Ham’s F-12培养基,pH 7.2)中孵育,该缓冲液含有200 pM 125I活化调节、正常T细胞表达和分泌(RANTES)、125I巨噬细胞炎性蛋白1α(MIP-1α)或125I-MIP-1β。结合反应在室温下进行40分钟。通过用冷磷酸盐缓冲盐水(PBS)洗涤两次无细胞配体来终止结合反应。用闪烁计数器 记录细胞相关放射性。以类似的方式进行了Cenicriviroc(CVC)/TAK-652对125I-RANTES与CCR1、125I-单核细胞趋化蛋白1(MCP-1)与CCR2b、125I-嗜酸性粒细胞趋化因子与CCR3、125I-胸腺和活化调节趋化因子(TARC)与CCR4以及125I-MIP-3β与CCR7结合的抑制作用的检测。 |
| 细胞实验 |
对小鼠单核细胞响应甲磺酸 Cenicriviroc (CVC) 治疗的迁移进行了三项独立评估。在雄性 C57BL/6 小鼠(n = 3;8-10 周龄)中,腹膜内注射巯基乙酸盐 (TG)。 48小时后,通过腹腔灌洗提取活化的巨噬细胞。将 1 μM 甲磺酸西尼基韦罗溶液添加到细胞中并孵育两小时。流式细胞仪通过分析从下室提取的细胞来计数 F4/80+CD11b+ 巨噬细胞的数量。使用 FlowJo 软件对结果进行分析[1]。
Cenicriviroc表型活性已使用PBMC表型敏感性试验对四种R5-、一种X4-和一种双嗜性HIV-2临床原发分离株进行了测试。所有分离物都是通过共培养PHA激活的PBMC获得的,这些PBMC来自法国HIV-2队列中不同的HIV-2感染的CCR5拮抗剂未成年患者,并且之前使用相同的方案进行了马拉韦敏感性测试。使用Ghost(3)细胞系通过表型分析确定HIV-2趋向性。结果:对于所测试的4株R5 HIV-2临床分离株,西尼利韦罗的有效浓度50%EC50分别为0.03、0.33、0.45和0.98 nM,与马拉韦罗的观察浓度相似:分别为1.13、0.58、0.48和0.68 nM。西尼利韦罗的最大抑制百分比(MPI)为94、94、93和98%,与马拉韦罗的观察结果相似(分别为93、90、82、100%)。双嗜性和X4嗜性HIV-2菌株对西尼利韦罗具有耐药性,EC50>1000 nM,MPI分别为33%和4%。 结论:在这项首次评估HIV-2对西尼利韦罗敏感性的研究中,我们观察到其对HIV-2 R5嗜性毒株的体外活性与马拉韦罗相似。因此,在有限的HIV-2治疗药物库中,西尼利韦可提供每日一次的治疗机会。有必要进行临床研究。[3] 雄性C57BL/6小鼠腹腔注射TG,48小时后腹腔灌洗收集活化的巨噬细胞。使用带有 5 μm 孔径聚碳酸酯过滤器的 Transwell1 室来测定趋化性。简而言之,细胞用 1 nM CCL2 和/或 1 μM Cenicriviroc(溶解在 0.5% 乙酸二甲亚砜中,并用无血清 Roswell Park Memorial Institute-1640 培养基和 0.5% 牛血清白蛋白按 1:1000 稀释)培养两小时)。使用 3 激光 BD FACSCanto,从下室提取细胞并进行流式细胞术分析,以计算 F4/80+CD11b+ 巨噬细胞的数量。使用FlowJo软件对结果进行分析。 |
| 动物实验 |
雄性C57BL/6小鼠(n = 44;8-10周龄)被分为5组,并于第1-5天进行灌胃给药(PO)。这些组包括:非疾病对照组、溶剂对照组(每日两次,BID)、塞尼西维罗甲磺酸盐5 mg/kg/天(CVC5,每日两次,BID)、塞尼西维罗甲磺酸盐20 mg/kg/天(CVC20,每日两次,BID)、塞尼西维罗甲磺酸盐100 mg/kg/天(CVC100,每日两次,BID)以及塞尼西维罗甲磺酸盐20 mg/kg/天(每日一次,QD)。除非疾病对照组外,所有组均在给药后两小时腹腔注射3.85%硫代乙醇酸盐(TG)(1 mL/只),以在第4天诱导腹膜炎[1]。
雄性 C57BL/6 小鼠(n = 44;8-10 周龄)分为以下几组,在第 1-5 天进行灌胃(PO)治疗:非疾病对照组、每日两次(BID)载体对照组、5 mg/kg/天(Cenicriviroc5)BID 组、20 mg/kg/天(Cenicriviroc20)BID 组、100 mg/kg/天(Cenicriviroc100)BID 组、Cenicriviroc20 QD 组和阳性对照组(1 mg/kg QD)——一种皮质类固醇,已证明可减轻多种动物模型中的炎症。在第4天,除非疾病对照组外,所有组均在给药后2小时腹腔注射3.85% TG(1 mL/只),以诱导腹膜炎。 在硫代乙醇酸盐诱导的小鼠腹膜炎模型中,评估了单核细胞/巨噬细胞的体内募集情况。在小鼠单核细胞上,评估了CCL2诱导的趋化作用。在硫代乙酰胺诱导的大鼠肝纤维化模型以及饮食诱导的非酒精性脂肪性肝炎(NASH)和肾纤维化小鼠模型中,评估了CVC的抗纤维化作用。研究评估指标包括体重和肝/肾重量、肝功能检查、肝/肾形态和胶原沉积、纤维化基因和蛋白表达以及药代动力学分析。 [1] 人体单剂量安全性和药代动力学研究。[5] 一项双盲 I 期临床试验旨在评估 TAK-652 单次口服给药在人体中的安全性、耐受性和药代动力学。本研究纳入 24 名健康志愿者(安慰剂组 2 名,各剂量组 6 名),受试者在空腹状态下口服三种剂量(25、50 和 100 mg)的 TAK-652 溶液。TAK-652 溶液的配制方法为:0.5% (w/v) 甲基纤维素、0.1% (w/v) 聚山梨醇酯 80 和 2 mM 盐酸溶于蒸馏水中。安慰剂溶液为0.5%(w/v)甲基纤维素、0.1%(w/v)聚山梨醇酯80和2 mM盐酸的蒸馏水溶液。剂量选择基于临床前药代动力学数据的异速缩放以及临床前毒理学研究结果(未观察到不良反应)。筛选在给药前3周进行,研究后评估在给药后5至7天进行。通过体格检查(筛选和研究结束后)、生命体征记录(筛选、给药前、给药后1、2、4、8和24小时以及研究结束后)、心电图(ECG;筛选、给药前、给药后2、6和24小时以及研究结束后)、临床实验室检查(血液学、血清化学和尿液分析;筛选、给药前、给药后24小时以及研究结束后)和不良事件记录(给药前、给药后3、12和24小时以及研究结束后)评估安全性和耐受性。采集系列血样以测定TAK-652的血浆浓度。血样采集时间点为给药前(0小时),随后在给药后0.5、1、2、3、4、6、8、12和24小时采集。样本立即进行处理,并采用液相色谱/串联质谱法定量测定血浆中TAK-652的浓度。血浆中TAK-652的定量下限为0.05 ng/ml。采用WinNonlin 3.2 Enterprise软件,通过非房室模型估算药代动力学参数。根据测得的浓度计算每位受试者的最大血浆浓度(Cmax)和达峰时间(Tmax)。采用梯形法则,根据测得的浓度计算每位受试者从零时点到最后一个可定量浓度的血浆浓度-时间曲线下面积(AUC0-tz)。 |
| 药代性质 (ADME/PK) |
在所有剂量下,TBR-652 的血浆峰浓度均在 3-4 小时内达到(表 2)。Helmert 对比显示,稳态浓度 (Css) 在第 8 天达到。对数正态 (ln) 转换后的 24 小时浓度-时间曲线下面积 (AUC0-24) 和血浆峰浓度 (Cmax) 表明,这些参数的增加幅度大于剂量依赖性。[1]
图 5 显示了给药后 30 分钟至 24 小时内各剂量 TAK-652 的平均血浆浓度。所有剂量下,药物在给药后 30 分钟和 24 小时均可在血浆中检测到。表 6 列出了健康志愿者单次口服给药后的估计药代动力学参数。总体而言,TAK-652 具有良好的口服吸收和较长的血浆半衰期。服用 25 毫克后 24 小时,其血浆浓度为 7.2 ng/ml,相当于 9.1 nM [5]。 |
| 毒性/毒理 (Toxicokinetics/TK) |
在所研究的剂量范围内,TBR-652总体上安全且耐受性良好。接受活性药物治疗的受试者中最常见的治疗期间出现的不良事件(AE)包括:胃肠道疾病(n = 19,43%);全身性疾病(n = 11,25%);神经系统疾病(n = 10,23%);呼吸系统、胸部和纵隔疾病(n = 10,23%);感染和寄生虫病(n = 7,16%);以及精神疾病(n = 5,11%)。接受TBR-652治疗的受试者中,大多数不良事件的严重程度为轻度(n = 24)或中度(n = 5)。仅有1例接受TBR-652治疗的受试者出现严重不良事件,即肩部脓肿,经研究者判断为非严重不良事件且与研究药物无关,无需治疗,且无后遗症。未发生危及生命的不良事件,未因不良事件而终止试验,也未发生死亡。所有不良事件均被判定为与研究药物明确相关。表3列出了报告为可能或很可能与研究药物相关的不良事件。大多数不良事件无需干预即可缓解。TBR-652组仅有4名受试者因评估为可能与研究药物相关的不良事件而需要同时服用其他药物。这些不良事件分别为:25 mg剂量组因腹痛服用氢可酮和对乙酰氨基酚;50 mg剂量组因头痛服用对乙酰氨基酚;100 mg剂量组因恶心服用昂丹司琼;以及150 mg剂量组一名受试者因头痛和恶心服用布洛芬。所有需要治疗的不良事件均被认为很可能与研究药物相关。[4]
24名接受治疗的受试者中,未发生因不良事件而退出试验的情况。共有4名受试者报告了6例临床不良事件。在六起不良事件中,两起与剂量无关的症状(头痛和疲劳)被认为可能与研究药物相关。其余四起轻微不良事件(头痛、鼻咽炎、感觉减退和眩晕)似乎与TAK-652的给药无关,但这一结论仍需进一步研究证实。研究期间,血清化学、血液学和尿液分析数据均未观察到与治疗或剂量相关的趋势。仰卧位收缩压、舒张压和脉率也未观察到与剂量相关的趋势。研究期间,所有受试者的生命体征或心电图均未观察到与治疗或剂量相关的明显趋势。尤其值得注意的是,在任何剂量的TAK-652下,均未发现QTc间期延长的证据。接受任何剂量的受试者,其心电图形态均未观察到具有临床意义的改变。研究结束后,未观察到具有临床意义的改变。因此,在健康男性受试者中,单次口服 TAK-652(25、50 和 100 mg 溶液)是安全且耐受性良好的。[5] |
| 参考文献 |
|
| 其他信息 |
塞尼克利维罗甲磺酸盐是塞尼克利维罗的甲磺酸盐形式,塞尼克利维罗是一种口服生物利用度高的双重抑制剂,可抑制人CC趋化因子受体2型(CCR2;CD192)和5型(CCR5;CD195),具有潜在的免疫调节、抗炎和抗病毒活性。口服后,塞尼克利维罗可特异性结合并抑制CCR2和CCR5的激活。这可抑制CCR2/CCR5介导的信号转导通路,并可能抑制炎症过程。G蛋白偶联趋化因子受体CCR2和CCR5表达于单核细胞和巨噬细胞表面,并刺激其迁移和浸润;它们在炎症和自身免疫性疾病中发挥关键作用。此外,Cenicriviroc 通过与 CCR5 辅助受体相互作用抑制人类免疫缺陷病毒 (HIV)-1 的入侵。
Cenicriviroc 属于苯并唑辛类化合物,其化学名称为 (5Z)-1,2,3,4-四氢-1-苯并唑辛,其 1、5 和 8 位分别被 2-甲基丙基、N-{4-[(S)-(1-丙基-1H-咪唑-5-基)甲亚磺酰基]苯基}甲酰胺基和 4-(2-丁氧基乙氧基)苯基取代。它是一种强效的趋化因子 2 和 5 受体拮抗剂,目前正在开发用于治疗成人非酒精性脂肪性肝炎 (NASH) 引起的肝纤维化。它具有趋化因子受体5拮抗剂、抗HIV药物、趋化因子受体2拮抗剂、抗风湿药和抗炎药的双重活性。它是一种二醚、咪唑类化合物、亚砜、芳香醚、仲酰胺和苯并唑辛类化合物。 Cenicriviroc已用于HIV感染/AIDS、AIDS痴呆综合征、非酒精性脂肪性肝炎、人类免疫缺陷病毒和HIV-1相关认知运动综合征的治疗试验。 Cenicriviroc是一种口服生物利用度高的双重抑制剂,可抑制人CC趋化因子受体2型(CCR2;CD192)和5型(CCR5;CD195),具有潜在的免疫调节、抗炎和抗病毒活性。口服后,cenicriviroc 可特异性结合并阻止 CCR2 和 CCR5 的激活。这抑制了 CCR2/CCR5 介导的信号转导通路,并可能抑制炎症过程。G 蛋白偶联趋化因子受体 CCR2 和 CCR5 表达于单核细胞和巨噬细胞表面,并刺激它们的迁移和浸润;它们在炎症和自身免疫性疾病中发挥关键作用。此外,cenicriviroc 还通过与 CCR5 共受体相互作用抑制人类免疫缺陷病毒 (HIV)-1 的进入。 另见:Cenicriviroc 甲磺酸盐(注释已移至)。 药物适应症 治疗非酒精性脂肪性肝炎 (NASH) Cenicriviroc 是一种 CCR5 拮抗剂,可阻止人类免疫缺陷病毒 1 型 (HIV-1) 进入细胞。 HIV-1包膜糖蛋白的CCR5结合区是中和抗体(NAbs)的重要靶点,因此,赋予cenicriviroc耐药性的突变可能会影响其对NAbs的敏感性。本研究利用体外诱导产生cenicriviroc或NAbs耐药的HIV-1变异株,探讨了cenicriviroc耐药性和NAbs耐药性之间的关系。cenicriviroc耐药变异株KK652-67(KK株在浓度递增的cenicriviroc存在下传代67次)对针对V3环、CD4诱导区(CD4i)和CD4结合位点(CD4bs)的NAbs敏感,而产生cenicriviroc耐药株KK652-67的野生型(WT)亲代HIV-1株KKWT则对这些NAbs耐药。 KK652-67 的 V3 区对于 cenicriviroc 耐药性至关重要,并且是 V3、CD4i 和 CD4bs 表位对中和抗体 (NAb) 高度敏感的关键因素。此外,从 cenicriviroc 耐药菌株 KK652-67 中诱导产生对 V3 NAb 0.5γ 和 CD4i NAb 4E9C 耐药的变异株,导致其表型恢复至与亲本菌株 KKWT 相似的 cenicriviroc 敏感表型。对 0.5γ 和 4E9C 的耐药性是由 V3 和 C3 区中位于赋予 cenicriviroc 耐药性的氨基酸替换附近的 R315K、G324R 和 E381K 这三个新的氨基酸替换引起的。重要的是,CCR5 结合区的这些氨基酸变化也是导致表型恢复至 cenicriviroc 敏感表型的原因。这些结果表明,存在一些关键氨基酸残基,使得对cenicriviroc的耐药性与对中和抗体的耐药性不相容。这意味着cenicriviroc和中和抗体可能限制彼此耐药变异株的出现。[2] 首个小分子CCR5拮抗剂TAK-779由于口服生物利用度低,未能被开发为抗人类免疫缺陷病毒(抗HIV-1)药物。TAK-652是TAK-779的口服生物利用度高的衍生物,具有强效的抗HIV-1活性。TAK-652在纳摩尔浓度下即可抑制RANTES(活化调节正常T细胞表达和分泌的分子)、巨噬细胞炎症蛋白1α(MIP-1α)和MIP-1β与表达CCR5的细胞的结合。 TAK-652 还能抑制单核细胞趋化蛋白 1 (MCP-1) 与表达 CCR2b 的细胞的结合。然而,其对配体与其他趋化因子受体结合的抑制作用有限。TAK-652 对使用 CCR5 受体的 (R5) HIV-1 病毒有效,但对使用 CXCR4 受体的 (X4) HIV-1 病毒完全无效。该化合物对含有逆转录酶和蛋白酶抑制剂耐药突变的 R5 HIV-1 临床分离株有效,其平均半数有效浓度 (EC50) 和 EC90 分别为 0.061 nM 和 0.25 nM。此外,携带不同亚型(A 至 G)包膜蛋白的重组 R5 病毒对 TAK-652 的敏感性相同。单次口服高达 100 mg 的 TAK-652 在人体中安全且耐受性良好。该化合物表现出良好的药代动力学特性,即使在服用 25 mg 后 24 小时,其血浆浓度仍为 7.2 ng/ml (9.1 nM)。因此,TAK-652 是一种很有前途的 HIV-1 新型进入抑制剂。[5] |
| 分子式 |
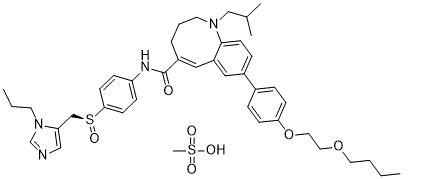
C42H56N4O7S2
|
|
|---|---|---|
| 分子量 |
793.05
|
|
| 精确质量 |
792.359
|
|
| 元素分析 |
C, 63.61; H, 7.12; N, 7.06; O, 14.12; S, 8.09
|
|
| CAS号 |
497223-28-6
|
|
| 相关CAS号 |
Cenicriviroc; 497223-25-3; 497223-22-0 (Cenicriviroc Sulfone)
|
|
| PubChem CID |
11285791
|
|
| 外观&性状 |
Light yellow to yellow solid powder
|
|
| tPSA |
168
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
10
|
|
| 可旋转键数目(RBC) |
17
|
|
| 重原子数目 |
55
|
|
| 分子复杂度/Complexity |
1150
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
S(CC1=CN=CN1CCC)(C1C=CC(=CC=1)NC(C1=CC2C=C(C3C=CC(=CC=3)OCCOCCCC)C=CC=2N(CCC1)CC(C)C)=O)=O.S(C)(=O)(=O)O |t:20|
|
|
| InChi Key |
IXPBPUPDRDCRSY-YLZLUMLXSA-N
|
|
| InChi Code |
InChI=1S/C41H52N4O4S.CH4O3S/c1-5-7-22-48-23-24-49-38-15-10-32(11-16-38)33-12-19-40-35(25-33)26-34(9-8-21-44(40)28-31(3)4)41(46)43-36-13-17-39(18-14-36)50(47)29-37-27-42-30-45(37)20-6-2;1-5(2,3)4/h10-19,25-27,30-31H,5-9,20-24,28-29H2,1-4H3,(H,43,46);1H3,(H,2,3,4)/b34-26+;/t50-;/m0./s1
|
|
| 化学名 |
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.5 mg/mL (3.15 mM) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶。
*生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (2.62 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (2.62 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.2610 mL | 6.3048 mL | 12.6095 mL | |
| 5 mM | 0.2522 mL | 1.2610 mL | 2.5219 mL | |
| 10 mM | 0.1261 mL | 0.6305 mL | 1.2610 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05630885 | Active Recruiting |
Drug: CVC 150 mg Drug: CVC 300 mg |
HIV-1-infection | National Institute of Allergy and Infectious Diseases (NIAID) |
May 30, 2023 | Phase 2 |
| NCT02684799 | Completed | Drug: Cenicriviroc Drug: Omeprazole Drug: Famotidine |
Healthy | Tobira Therapeutics, Inc. | January 31, 2016 | Phase 1 |
| NCT02342067 | Completed | Drug: Cenicriviroc Drug: Pioglitazone |
Healthy | Tobira Therapeutics, Inc. | December 2014 | Phase 1 |
| NCT02685462 | Completed | Drug: Rosuvastatin Drug: Atorvastatin Drug: Simvastatin Drug: Digoxin Drug: Caffeine |
Healthy | Tobira Therapeutics, Inc. | January 31, 2016 | Phase 1 |
| NCT04500418 | Terminated | Drug: Cenicriviroc (CVC) Drug: Placebo |
Covid19 | Charite University, Berlin, Germany |
August 25, 2020 | Phase 2 |
 Inhibitory effect of TAK-652 on binding of RANTES (A), MIP-1α (B), and MIP-1β (C) to CCR5.Antimicrob Agents Chemother.2005 Nov;49(11):4584-91. |
|---|
 Inhibitory effect of TAK-652 on ligand binding to various chemokine receptors.CHO cells expressing CCR1 (open circles), CCR2b (open squares), CCR3 (filled triangles), CCR4 (open triangles), or CCR7 (filled circles) were incubated with various concentrations of TAK-652 in binding buffer containing125I-labeled RANTES, MCP-1, eotaxin, TARC, or MIP-3β, respectively. Binding reactions were performed at room temperature and terminated by washing out the cell-free ligand with PBS. The cell-associated radioactivity was measured with a scintillation counter. Data represent means ± standard deviations for triplicate wells.Antimicrob Agents Chemother.2005 Nov;49(11):4584-91. |
 Antiviral activity of TAK-652 against R5X4 HIV-1 in U87.CD4.CCR5 and U87.CD4.CXCR4 cells. The cells were infected with R5X4 HIV-1 (HE) and incubated in the presence of test compounds (100 nM). After incubation for 6 h, the cells were washed to remove unadsorbed viral particles and further incubated in the presence of the same concentration of the test compounds for 3 days.
|
 HGS004
HGS004
 CCR3 antagonist 2
CCR3 antagonist 2
 LMD-902
LMD-902
 RAP-103
RAP-103
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 Plasma concentration-time profiles after single oral administration of TAK-652 to humans.
Plasma concentration-time profiles after single oral administration of TAK-652 to humans. 463611831
463611831
