| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
GSK-3β (IC50 = 0.58 nM); GSK-3α (IC50 = 0.65 nM); cdc2 (IC50 = 3700 nM)
Glycogen Synthase Kinase 3 (GSK3), including both GSK3α and GSK3β isoforms. For GSK3β, the IC₅₀ value was determined to be ~0.7 nM; for GSK3α, the IC₅₀ value was ~0.6 nM. The compound exhibited high selectivity for GSK3: it showed minimal inhibitory activity against other kinases, with IC₅₀ values >100 nM for cyclin-dependent kinase 2 (CDK2), extracellular signal-regulated kinase 2 (ERK2), and protein kinase B (Akt) [2] |
|---|---|
| 体外研究 (In Vitro) |
CHIR-98014 抑制人 GSK-3β,Ki 为 0.87 nM。 CHIR-98014 对于预防小鼠和大鼠 GSK-3 非常有效。尽管 CHIR-98014 作为 ATP 结合的直接竞争性抑制剂,但与 20 种其他蛋白激酶(例如 Cdc2、ERK2、Tie-2 和 KDR)相比,CHIR-98014 对 GSK-3 的选择性高出 500 倍至 >1000 倍。 CHIR-98014 抑制 Cdc2 的 IC50 为 3.7 M。值得注意的是,CHIR 98014 仅区分 GSK-3 及其最接近的同源物 CDC2 和 ERK2,尽管它对 GSK-3 的高度同源性和亚型表现出相似的效力。当抑制剂 CHIR98014 以不断增加的浓度应用于表达胰岛素受体的 CHO-IR 细胞或原代大鼠肝细胞时,会产生高于基础值 2 至 3 倍的 GS 活性比刺激。对于大鼠肝细胞和 CHO-IR,引起半最大 GS 刺激 (EC50) 的 CHIR-98014 浓度为 107 nM。[1]
1. 在分化的L6大鼠骨骼肌肌管细胞中,用CHIR-98014(0.1 nM-1 μM,处理24小时)处理后,通过[¹⁴C]-葡萄糖掺入法检测发现,该药物呈剂量依赖性促进糖原合成。在10 nM浓度下,糖原合成较溶剂对照组增加约2.3倍。此效应与糖原合成酶(GS)在Ser⁶⁴¹位点的磷酸化水平升高(该位点磷酸化抑制GS活性,去磷酸化则激活)相关,通过Western blot可检测到这一变化。此外,CHIR-98014(10 nM)可增强胰岛素诱导的糖原合成:在1 nM胰岛素存在时,该药物可使糖原积累量较单独使用胰岛素时再增加约1.8倍 [2] 2. 在3T3-L1小鼠脂肪细胞中,CHIR-98014(1 nM-100 nM,处理16小时)通过[³H]-2-脱氧葡萄糖摄取实验检测显示,可促进葡萄糖转运。在10 nM浓度下,葡萄糖转运较对照组增加约1.9倍。该药物还能增强胰岛素刺激的葡萄糖转运:与10 nM胰岛素联合使用时,葡萄糖摄取量较单独使用胰岛素时高约2.5倍。Western blot分析表明,CHIR-98014(10 nM)可增加Akt在Ser⁴⁷³位点的磷酸化(胰岛素信号通路的下游效应分子),并降低GSK3β在Ser⁹位点的磷酸化(GSK3β抑制的标志物) [2] 3. 在转染了β-连环蛋白响应性荧光素酶报告基因(TOPFlash)的HEK293细胞中,CHIR-98014(0.1 nM-1 μM)呈剂量依赖性激活Wnt/β-连环蛋白通路,EC₅₀约为2 nM。在10 nM浓度下,荧光素酶活性较对照组高约8倍,同时伴随β-连环蛋白在细胞核内的积累(免疫荧光染色) [2] |
| 体内研究 (In Vivo) |
GSK-3 抑制剂 CHIR-98014 可激活胰岛素敏感瘦 Zucker 大鼠和胰岛素抵抗 ZDF 大鼠分离的 I 型骨骼肌中的 GS 活性比。从 ZDF 大鼠中分离的比目鱼肌显示出对激活 GS 的胰岛素具有显着抵抗性,但对 500 nM CHIR-98014 的反应程度与来自瘦 Zucker 大鼠的肌肉相同程度(增加 40%)。值得注意的是,胰岛素加 CHIR-98014 的 GS 激活对瘦 Zucker 大鼠的肌肉具有附加作用,并且比 ZDF 大鼠的肌肉具有更大的附加作用。这些细胞和肌肉中的总 GS 活性不会被 CHIR-98014 或胰岛素改变。同时,CHIR-98014 不会影响瘦动物肌肉中的胰岛素剂量反应。高血糖的降低和葡萄糖处理的改善不仅限于db/db小鼠和ZDF大鼠,在ob/ob小鼠、饮食诱导的糖尿病C57BL/6小鼠和用CHIR-治疗的葡萄糖不耐症SHHF大鼠中也观察到类似的结果。 98014。此外,CHIR-98014 还可降低出生后大鼠皮质和海马中 tau 蛋白的磷酸化 (Ser396)。
1. 在雄性ob/ob小鼠(2型糖尿病遗传模型,8-10周龄)中,口服给予CHIR-98014(1 mg/kg、3 mg/kg、10 mg/kg,每日1次,连续7天),呈剂量依赖性降低空腹血糖(FBG)。在10 mg/kg剂量下,第7天的FBG从溶剂组的24.3 mM降至15.7 mM(降低约35%)。该药物还能增加肝脏和骨骼肌中的糖原含量:在10 mg/kg剂量下,肝糖原较对照组高约2.1倍,腓肠肌糖原较对照组高约1.8倍 [2] 2. 在雄性db/db小鼠(另一种2型糖尿病模型)中,单次口服CHIR-98014(10 mg/kg)后,餐后血糖(PBG)在给药2小时时降低约40%,效应可持续长达6小时。胰岛素耐量试验(ITTs)显示,CHIR-98014(3 mg/kg,口服3天)可改善胰岛素敏感性:ITT期间的血糖曲线下面积(AUC)较溶剂组降低约25% [2] |
| 酶活实验 |
聚丙烯 96 孔板每孔填充 300 μL 含有激酶的缓冲液(50 mM tris HCl、10 mM MgCl2、1 mM EGTA、1 mM 二硫苏糖醇、25 mM β-甘油磷酸、1 mM NaF、0.01% BSA,pH 7.5) 、肽底物和任何活化剂。在所有无细胞测定中,将 CHIR-98014 或对照添加到 3.5 μL DMSO 中,然后添加 50 μL ATP 库存,以产生 1 μM ATP 的终浓度。孵育后,将一式三份的 100 L 等分试样添加到 Combiplate 8 板中,其中每孔含有 100 µL 浓度的 50 mM ATP 和 20 mM EDTA。将孔用 PBS 冲洗五次,填充 200 L 闪烁液,密封,放置 30 分钟,然后在第一小时后在闪烁计数器中计数。整个过程在室温下进行。
1. GSK3β激酶活性检测:将重组人GSK3β(5 ng)与合成肽底物(序列:YRRAAVPPSPSLSRHSSPHQpSEDEEE,50 μM)在含20 mM Tris-HCl(pH 7.5)、10 mM氯化镁(MgCl₂)、1 mM二硫苏糖醇(DTT)和10 μM [γ-³³P]-ATP的反应缓冲液中孵育。加入CHIR-98014(0.01 nM-1 μM)后,混合物在30°C孵育60分钟。取20 μL反应液点样于磷酸纤维素纸上终止反应,用1%磷酸洗涤3次以去除未掺入的[γ-³³P]-ATP。通过液体闪烁计数测定放射性,根据剂量-反应曲线(抑制率vs.对数浓度)计算IC₅₀ [2] 2. GSK3α激酶活性检测:实验流程与GSK3β检测一致,仅替换为重组人GSK3α(5 ng)。使用相同剂量范围的CHIR-98014和数据分析方法确定GSK3α的IC₅₀ [2] 3. 激酶选择性检测:对于其他激酶(CDK2、ERK2、Akt),将重组酶(5-10 ng)与其各自的肽底物、[γ-³³P]-ATP和CHIR-98014(0.1 nM-10 μM)在激酶特异性反应缓冲液中孵育。按上述方法测定放射性,计算IC₅₀以评估选择性 [2] |
| 细胞实验 |
表达人胰岛素受体的 CHO-IR 细胞在含有 10% 胎牛血清且不含次黄嘌呤的 Hamm's F12 培养基中生长至 80% 汇合。将胰蛋白酶处理的细胞以 1 × 106 个细胞/孔的密度接种在 2 mL 不含胎牛血清的培养基中的 6 孔板中。 24 小时后,用 1 mL 含有 GSK-3 抑制剂 CHIR 98014 或对照(最终 DMSO 浓度 0.1%)的无血清培养基替换培养基,在 37 °C 下培养 30 分钟。将细胞在含有 1 mM EDTA、1 mM DTT、100 mM NaF、1 mM 苯甲基磺酰氟和 25 g/mL 亮抑肽素(缓冲液 A)的 50 mM tris (pH 7.8) 中冷冻/解冻,并在 30 ℃ 离心 15 分钟。 4°C/14000 克。使用不存在时的 GS 活性来计算 GS 的活性比。
1. L6肌管细胞糖原合成实验:将L6细胞接种于24孔板,在含2%马血清的DMEM中培养7天分化为肌管细胞。肌管细胞饥饿血清16小时后,用CHIR-98014(0.1 nM-1 μM)±胰岛素(1 nM)处理24小时。在处理的最后4小时加入[¹⁴C]-葡萄糖(0.5 μCi/mL)。用冷PBS洗涤细胞,用10%三氯乙酸(TCA)裂解细胞,糖原在4°C沉淀过夜。沉淀的糖原用乙醇洗涤,溶解于水中,通过液体闪烁计数测定放射性,以量化糖原合成 [2] 2. 3T3-L1脂肪细胞葡萄糖转运实验:3T3-L1前脂肪细胞通过胰岛素、地塞米松和IBMX处理8天分化为脂肪细胞。脂肪细胞饥饿血清4小时后,用CHIR-98014(1 nM-100 nM)±胰岛素(10 nM)处理16小时。加入[³H]-2-脱氧葡萄糖(0.1 μCi/mL)孵育10分钟,用含200 μM根皮素(抑制葡萄糖转运体)的冷PBS洗涤细胞。用0.1% SDS裂解细胞,测定放射性以确定葡萄糖摄取量 [2] 3. β-连环蛋白报告基因实验:使用转染试剂将TOPFlash(β-连环蛋白响应性荧光素酶质粒)和pRL-TK(海肾荧光素酶质粒,内参对照)转染至HEK293细胞。转染24小时后,用CHIR-98014(0.1 nM-1 μM)处理细胞24小时。采用双荧光素酶报告基因检测系统测定荧光素酶活性,将萤火虫荧光素酶活性归一化为海肾荧光素酶活性 [2] |
| 动物实验 |
药物及给药方法[2]
SB216763 (30 mg kg−1) 和 CHIR98014 (30 mg kg−1) 用 DMSO 重悬后静脉注射;AR-A014418 (30 mg kg−1) 溶于 100% PEG400 后口服;靛玉红-3′-单肟 (20 mg kg−1) 和阿斯特帕隆 (20 mg kg−1) 溶于 20% DMSO/25% Tween-80 溶液中,分别腹腔注射和皮下注射。所有药物研究均使用同一窝的 P12 大鼠进行。对照组动物给予相应的溶剂,两组动物分别在给药后 1、2 和 4 小时处死,用于脑组织暴露量测定(见下一节)、蛋白质印迹分析和 GSK-3β 活性测定。为评估每种化合物的功效,实验至少重复三次,并根据脑暴露数据确定时间点。将氯化锂(100 和 200 mg kg−1)溶于无菌水中,经口给予动物。注射后 8 小时处死 P12 大鼠。部分同窝大鼠作为对照组,经口给予溶于无菌水中的氯化钠(100 或 200 mg kg−1)。[2] 脑暴露量测定[2] 采用湍流色谱法 (HTLC) 结合串联质谱法 (MS/MS) 分析大鼠脑匀浆中 SB216763、靛玉红-3′-单肟、阿斯特帕隆、CHIR98014 和 AR-A014418 的暴露水平。向样品中加入四倍量的 70% (v/w) 乙腈,并在自动匀浆机中进行匀浆。将脑匀浆在 5 °C 下以 6000 g 离心 15 分钟,并分析上清液。使用未经处理的大鼠脑匀浆制备每种化合物的校准曲线(1–1000 ng ml⁻¹ 脑匀浆)。向 25 μl 脑匀浆或校准标准品中加入 25 μl 含内标(西酞普兰)的 10% 甲醇溶液,然后在 5 °C 下以 6000 g 离心 20 分钟。使用 HTS PAL 自动进样器将 10 μl 样品注入 HTLC 系统。使用 Cyclone HTLC 色谱柱(0.5 × 50 mm,50 μm),以 0.1% 甲酸水溶液为流动相,以 2 ml min⁻¹ 的流速纯化含有 AR-A014418 的样品 15 秒。将化合物从高通量色谱 (HTLC) 中提取出来,方法是在进样环中加入 100 μL 0.1% 甲酸/90% 乙腈溶液,然后转移至分析柱 X-Terra MS C8 (20 × 2.1 mm, 3.5 μm),流动相为 0.1% 甲酸水溶液,在 120 秒内(流速 0.08 mL min⁻¹)。之后,使用 0.1% 甲酸/2% 乙腈至 0.1% 甲酸/98% 乙腈的梯度洗脱液,洗脱时间为 45 秒,最后用 0.1% 甲酸/98% 乙腈溶液洗脱 120 秒(流速 0.5 mL min⁻¹)。化合物的检测采用 Ultima 三重四极杆质谱仪 (Waters),并使用多反应监测 (MRM) 正离子模式,在最佳条件下进行。对于 AR-A014418,使用了 308.9 → 121.7 的转换。 通过在利多卡因麻醉的尾尖处进行浅剪取血。直接测量血糖,或收集肝素化血浆用于测量葡萄糖或胰岛素。动物预先采血,并随机分配到载体对照组或 GSK-3 抑制剂治疗组。对于葡萄糖耐量试验 (GTT),动物在整个试验过程中禁食,并在首次预采血前 3 小时(db/db 小鼠)或采血前 16 小时(ZDF 大鼠)的前一天晚上停止喂食。在测定禁食 ZDF 大鼠血浆葡萄糖和胰岛素变化的时间进程时,在给予试验药物前 16 小时停止喂食。GS 活性比值计算为在 5 mM 葡萄糖-6-磷酸存在下的 GS 活性除以在没有葡萄糖-6-磷酸存在下的活性。 1. ob/ob小鼠慢性治疗方案:雄性ob/ob小鼠(8-10周龄,体重40-45 g)随机分为4组(每组n=6):赋形剂组(0.5%甲基纤维素)、CHIR-98014组(1 mg/kg、3 mg/kg、10 mg/kg)。药物悬浮于0.5%甲基纤维素溶液中,每日一次灌胃给药,连续7天。小鼠在第0天(基线)和第7天进行空腹血糖(FBG)测定(尾静脉采血,血糖仪),测定前禁食6小时。第7天处死小鼠;取肝脏(左叶)和腓肠肌,液氮速冻,-80℃保存。采用比色法(糖原水解为葡萄糖,葡萄糖氧化酶检测)测定组织中糖原含量[2] 2. db/db小鼠急性降血糖方案:雄性db/db小鼠(10-12周龄)单次口服CHIR-98014(10 mg/kg,溶于0.5%甲基纤维素)或溶剂对照。分别于给药后0、1、2、4、6小时(进食状态)测量血糖(PBG)。对于胰岛素耐量试验(ITT),小鼠口服CHIR-98014(3 mg/kg)或溶剂对照3天,然后禁食4小时。腹腔注射胰岛素(0.75 U/kg),分别于注射胰岛素后0、15、30、60、120分钟测量血糖,计算血糖曲线下面积(AUC)[2]。 |
| 药代性质 (ADME/PK) |
1. 在雄性 CD-1 小鼠中,口服 CHIR-98014 (10 mg/kg) 的口服生物利用度约为 30%。血浆峰浓度 (Cₘₐₓ) 约为 85 ng/mL,在给药后约 1 小时 (Tₘₐₓ) 达到。消除半衰期 (t₁/₂) 约为 2.1 小时。组织分布分析显示,给药后 1 小时,药物在肝脏(峰浓度约为 240 ng/g)和骨骼肌(峰浓度约为 120 ng/g)中蓄积 [2]
|
| 毒性/毒理 (Toxicokinetics/TK) |
1. 在CD-1小鼠的急性毒性研究中,单次口服剂量高达100 mg/kg的CHIR-98014,7天内未观察到死亡或明显的毒性症状(例如嗜睡、共济失调)。药物组和溶媒组的体重增加相似[2]
2. 在ob/ob小鼠的7天慢性毒性研究中(1-10 mg/kg,口服),与溶媒组相比,CHIR-98014对血清丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)、肌酐或尿素氮(肝肾功能标志物)均无显著影响[2] 3. CHIR-98014在小鼠、大鼠和人血浆中的血浆蛋白结合率约为95%(通过平衡透析法测定)[2] |
| 参考文献 | |
| 其他信息 |
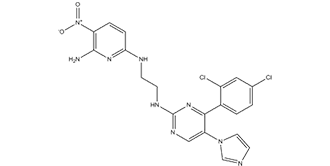
CHIR-98014 属于氨基嘧啶类化合物,其嘧啶环上分别在 2、4 和 5 位被 {2-[(6-氨基-5-硝基吡啶-2-基)氨基]乙基}氨基、2,4-二氯苯基和 1H-咪唑-1-基取代。它是一种强效的 ATP 竞争性 GSK3α 和 GSK3β 抑制剂(IC50 值分别为 0.65 nM 和 0.58 nM)。它具有多种活性,包括 EC 2.7.11.26(tau 蛋白激酶)抑制剂、细胞凋亡诱导剂、抗肿瘤剂、降血糖剂、Wnt 信号通路激活剂和 tau 蛋白聚集抑制剂。它是一种仲氨基化合物、二氯苯、咪唑类化合物、二氨基吡啶、氨基嘧啶和C-硝基化合物。
1. CHIR-98014 主要通过抑制 GSK3 发挥其抗糖尿病作用:抑制 GSK3 可降低 GS 的磷酸化(激活 GS 以促进糖原合成),并通过增加 Akt 磷酸化增强胰岛素信号传导,从而改善肌肉和脂肪组织中的葡萄糖摄取和糖原储存[2] 2. CHIR-98014 激活 Wnt/β-catenin 通路(通过稳定 β-catenin)提示可能存在脱靶效应,但在糖尿病模型中,其主要的治疗作用是通过抑制代谢组织(肌肉、肝脏、脂肪组织)中的 GSK3 介导的[2] 3. CHIR-98014 显示出优势与早期的 GSK3 抑制剂(例如 SB216763)相比,该药物具有更高的效力(对 GSK3 的 IC₅₀ 值更低)以及对 GSK3 相对于其他激酶的选择性,从而降低了脱靶毒性的风险 [2] |
| 分子式 |
C20H17CL2N9O2
|
|---|---|
| 分子量 |
486.3141
|
| 精确质量 |
485.088
|
| 元素分析 |
C, 49.39; H, 3.52; Cl, 14.58; N, 25.92; O, 6.58
|
| CAS号 |
252935-94-7
|
| 相关CAS号 |
556813-39-9 (CHIR98024);252935-94-7 (CHIR98014);CHIR98014 HCl;
|
| PubChem CID |
53396311
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.6±0.1 g/cm3
|
| 沸点 |
839.0±75.0 °C at 760 mmHg
|
| 闪点 |
461.2±37.1 °C
|
| 蒸汽压 |
0.0±3.1 mmHg at 25°C
|
| 折射率 |
1.753
|
| LogP |
3.76
|
| tPSA |
158.85
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
645
|
| 定义原子立体中心数目 |
0
|
| SMILES |
ClC1C([H])=C(C([H])=C([H])C=1C1C(=C([H])N=C(N=1)N([H])C([H])([H])C([H])([H])N([H])C1C([H])=C([H])C(=C(N([H])[H])N=1)[N+](=O)[O-])N1C([H])=NC([H])=C1[H])Cl
|
| InChi Key |
MDZCSIDIPDZWKL-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C20H17Cl2N9O2/c21-12-1-2-13(14(22)9-12)18-16(30-8-7-24-11-30)10-27-20(29-18)26-6-5-25-17-4-3-15(31(32)33)19(23)28-17/h1-4,7-11H,5-6H2,(H3,23,25,28)(H,26,27,29)
|
| 化学名 |
6-N-[2-[[4-(2,4-dichlorophenyl)-5-imidazol-1-ylpyrimidin-2-yl]amino]ethyl]-3-nitropyridine-2,6-diamine
|
| 别名 |
CT-98014; CT 98014; CT98014; CHIR 98014; CHIR-98014; 252935-94-7; 6-N-[2-[[4-(2,4-dichlorophenyl)-5-imidazol-1-ylpyrimidin-2-yl]amino]ethyl]-3-nitropyridine-2,6-diamine; CT-98014; 2,6-PYRIDINEDIAMINE, N6-[2-[[4-(2,4-DICHLOROPHENYL)-5-(1H-IMIDAZOL-1-YL)-2-PYRIMIDINYL]AMINO]ETHYL]-3-NITRO-; CHEMBL3185148; N2-(2-((4-(2,4-dichlorophenyl)-5-(1H-imidazol-1-yl)pyrimidin-2-yl)amino)ethyl)-5-nitropyridine-2,6-diamine; N6-[2-[[4-(2,4-DICHLOROPHENYL)-5-(1H-IMIDAZOL-1-YL)-2-PYRIMIDINYL]AMINO]ETHYL]-3-NITRO-2,6-PYRIDINEDIAMINE; CHIR-98014; CHIR98014;CHIR98014 HCl; CHIR-98014 hydrochloride
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~8 mg/mL (~16.5 mM)
Water: <1 mg/mL Ethanol: <1 mg/mL |
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0563 mL | 10.2815 mL | 20.5630 mL | |
| 5 mM | 0.4113 mL | 2.0563 mL | 4.1126 mL | |
| 10 mM | 0.2056 mL | 1.0282 mL | 2.0563 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
The aminopyrimidine GSK-3 inhibitor, CHIR98014, reduced tau phosphorylation in vivo.Br J Pharmacol.2007Nov;152(6):959-79. |
CHIR98014, reduced tau phosphorylation in a dose-dependent manner in vivo.Br J Pharmacol.2007Nov;152(6):959-79. |
Characterization of the postnatal rat model.Br J Pharmacol.2007Nov;152(6):959-79.Characterization of the postnatal rat model. Br J Pharmacol. 2007 Nov;152(6):959-79. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA



 463611831
463611831
