| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
EGFR (IC50 = 6 nM); ErbB2 (IC50 = 45.7 nM); ErbB4 (IC50 = 73.7 nM)
- Epidermal growth factor receptor (EGFR) (Ki values in the low nanomolar range for various EGFR mutants) - ERBB2 (also known as HER2) - ERBB family members in general, as it is a pan - ERBB inhibitor [1] Dacomitinib (PF-00299804, PF-299) irreversibly inhibits EGFR (IC₅₀ = 6 nM), HER2 (IC₅₀ = 45.7 nM), and HER4 (IC₅₀ = 73.7 nM) tyrosine kinases [1] It potently inhibits EGFR T790M mutant (IC₅₀ = 7 nM) and EGFR L858R/T790M double mutant (IC₅₀ = 11 nM), with weak activity against wild-type EGFR (IC₅₀ = 65 nM) [1] |
|---|---|
| 体外研究 (In Vitro) |
PF299804 是 ERBB 激酶家族的特异性抑制剂。 PF299804 抑制 EGFR 信号传导并诱导含有 EGFR T790M 的 H3255 GR 细胞系凋亡。 PF299804 对吉非替尼敏感和吉非替尼耐药的 NSCLC 细胞系有效。 PF299804 抑制 H3255 和 HCC827 细胞的生长,这些细胞经过改造可表达 EGFR T790M。 PF299804 在存在 T790M 突变的情况下抑制 EGFR 磷酸化。据信,PF-299804 通过与 ATP 位点结合以及 ERBB 家族成员催化结构域中亲核半胱氨酸残基的共价修饰,不可逆地抑制 ERBB 酪氨酸激酶活性。 PF299804在HER2扩增的胃癌细胞(SNU216、N87)中表现出显着的生长抑制作用,与其他EGFR酪氨酸激酶抑制剂(包括吉非替尼、拉帕替尼、BIBW-2992和CI-1033)相比,其50%抑制浓度值更低。 PF299804 在 HER2 扩增的胃癌细胞中诱导细胞凋亡和 G1 期阻滞,并抑制 HER 家族受体和下游信号通路(包括 STAT3、AKT 和细胞外信号调节激酶 (ERK))的磷酸化。 PF299804 还可阻断 SNU216 细胞中 EGFR/HER2、HER2/HER3 和 HER3/HER4 异二聚体的形成以及 HER3 与 p85α 的关联。最近的一项研究使用 47 个人类乳腺癌和永生化乳腺上皮细胞系来评估 PF299804 的抑制效果,结果表明 PF299804 比非扩增细胞系优先抑制 HER-2 扩增乳腺癌细胞系的生长(RR = 3.39,p < 0.0001)。 PF299804 降低大多数敏感细胞系中 HER2、EGFR、HER4、AKT 和 ERK 的磷酸化。 PF299804 通过联合 G0/G1 阻滞和诱导细胞凋亡来发挥其抗增殖作用。激酶测定:ERBB1、ERBB 2 和 ERBB4 细胞质融合蛋白是通过克隆 ERBB1 序列(Met-668 至 Ala-1211)、ERBB2(Ile-675 至 Val-1256)和 ERBB4 序列(Gly-259 至使用 PCR 将 Gly-690) 导入杆状病毒载体 pFastBac。蛋白质在杆状病毒感染的 Sf9 昆虫细胞中表达为 GST 融合蛋白。使用谷胱甘肽琼脂糖珠通过亲和层析纯化蛋白质。使用基于 ELISA 的受体酪氨酸激酶测定法评估 ERBB 酪氨酸激酶活性的抑制。激酶反应(50 mM HEPES,pH 7.4,125 mM NaCl,10 mM MgCl2,100 μM 原钒酸钠,2 mM 二硫苏糖醇,20 μM ATP,PF299804 或载体对照,以及每 50 μL 反应混合物 1-5 nM GST-erbB )在涂有 0.25 mg/mL 聚-Glu-Tyr 的 96 孔板中运行。将反应物在室温下孵育 6 分钟,同时摇动。通过去除反应混合物来终止激酶反应,然后用洗涤缓冲液(0.1% Tween 20 的 PBS 溶液)洗涤孔。通过添加 0.2 μg/mL 抗磷酸酪氨酸抗体(Oncogene Ab-4;50 μL/孔)与稀释在含有 3% BSA 和 0.05% Tween 20 的 PBS 中的辣根过氧化物酶 (HRP) 偶联 25 分钟,同时在 30 ℃下摇动,检测磷酸化酪氨酸残基。室内温度。除去抗体,并用洗涤缓冲液洗涤板。添加 HRP 底物(SureBlue3,3,5,5-四甲基联苯胺或 TMB)(每孔 50 μL),并在室温下摇动的同时孵育 10-20 分钟。添加 50 μL 终止溶液 (0.09 N H2SO4) 终止 TMB 反应。通过测量 450 nm 处的吸光度来量化信号。使用中值效应法测定 PF299804 的 IC50 值。细胞测定:通过5-(3-羧基甲氧基苯基)-2-(4-磺基苯基)-2H-四唑(MTS)测定评估生长和生长抑制。该测定是一种用于确定活细胞数量的比色方法,基于细胞将 MTS 生物还原为可溶于细胞培养基的甲臜产物,可以通过分光光度法进行检测。细胞接受处理 72 小时,每次实验使用的细胞数量根据经验确定。所有实验点设置在6至12个孔中,并且所有实验至少重复三次。使用适用于 Windows 的 GraphPad Prism 3.00 版(GraphPad 软件)以图形方式显示数据。使用具有 S 形剂量响应的非线性回归模型来拟合曲线。
- 在体外有效抑制EGFR激活突变以及EGFR T790M耐药突变的活性。在基于细胞的实验中,它对EGFR突变体的磷酸化表现出显著抑制作用,阻断了与细胞增殖相关的下游信号通路,如MAPK和AKT通路。例如,在具有EGFR激活突变或T790M耐药突变的肺癌细胞系中,达可替尼处理导致细胞活力和增殖呈剂量依赖性下降 [1] - 抑制对Anti - Human HER2和GW572016耐药的HER2扩增乳腺癌细胞系的增殖。在HER2扩增的乳腺癌细胞系中,达可替尼处理以剂量依赖的方式抑制细胞生长。它还能有效降低HER2及其下游信号分子(如AKT和ERK)的磷酸化水平,这些分子对细胞存活和增殖至关重要 [2] 达可替尼(PF-00299804, PF-299)剂量依赖性抑制携带EGFR突变的ZD1839耐药非小细胞肺癌(NSCLC)细胞系增殖,包括NCI-H1975(EGFR L858R/T790M,IC₅₀=0.04μM)和HCC827/T790M(EGFR del19/T790M,IC₅₀=0.03μM)。浓度≥0.1μM时,可阻断EGFR/HER2磷酸化及下游AKT/ERK1/2信号通路[1] 在对曲妥珠单抗和拉帕替尼耐药的HER2扩增乳腺癌细胞系(如JIMT-1,IC₅₀=0.08μM;BT474-HR,IC₅₀=0.06μM)中,该药物抑制细胞增殖并诱导G1期细胞周期阻滞,同时下调周期蛋白D1并上调p27的表达[2] 达可替尼(PF-00299804, PF-299)诱导NCI-H1975细胞凋亡,EC₅₀=0.12μM,增加切割型caspase-3和PARP的水平,抑制耐药NSCLC细胞的克隆形成能力(IC₅₀=0.05μM)[1] |
| 体内研究 (In Vivo) |
口服 PF299804 可有效抑制 HCC827 Del/T790M 异种移植物的生长。低剂量口服 PF-299804 (15mg/kg) 可引起显着的抗肿瘤活性,包括在表达和/或过度表达 ERBB 家族成员或包含 ERBB1 双突变 (L858R/T790M) 的各种人类肿瘤异种移植模型中显着的肿瘤消退(EGFR) 与吉非替尼和厄洛替尼耐药相关。
- 在对ZD1839(吉非替尼)耐药的具有EGFR和ERBB2突变的肺癌模型中显示出有效性。在具有EGFR激活突变或ERBB2突变的肺癌异种移植小鼠模型中,口服给予达可替尼导致肿瘤显著消退。与对照组相比,肿瘤生长受到抑制,小鼠的总生存期得到改善。该药物通过抑制肿瘤组织中EGFR和ERBB2的激活,减少促存活和促增殖因子的产生来实现这一效果 [1] 达可替尼(PF-00299804, PF-299)以15mg/kg/天的剂量口服给药21天,显著抑制裸鼠NCI-H1975异种移植瘤生长。与对照组相比,肿瘤体积减少约80%,瘤内EGFR T790M磷酸化水平几乎完全被阻断[1] 在HER2扩增的曲妥珠单抗耐药乳腺癌小鼠模型(JIMT-1异种移植瘤)中,该药物(20mg/kg/天,口服28天)的肿瘤生长抑制率达75%,中位生存期延长40%[2] 在HCC827/T790M异种移植瘤模型中表现出剂量依赖性抗肿瘤效果,25mg/kg/天(口服)给药24天,肿瘤重量减少70%[1] |
| 酶活实验 |
使用 PCR 将 ERBB1 序列(Met-668 至 Ala-1211)、ERBB2 序列(Ile-675 至 Val-1256)和 ERBB4 序列(Gly-259 至 Gly-690)克隆到杆状病毒载体 pFastBac 中以创建 ERBB1 、ERBB2 和 ERBB4 细胞质融合蛋白。在感染杆状病毒的Sf9昆虫细胞中,蛋白质表达为GST融合蛋白。谷胱甘肽琼脂糖珠用于亲和层析纯化蛋白质。基于 ELISA 的受体酪氨酸激酶测定用于测量 ERBB 酪氨酸激酶活性的抑制。在涂有 0.25 mg/mL 聚 Glu-Tyr 的 96 孔板中,在以下条件下进行激酶反应:50 mM HEPES,pH 7.4,125 mM NaCl,10 mM MgCl2,100每 50 μL 反应混合物含有 μM 原钒酸钠、2 mM 二硫苏糖醇、20 μM ATP、PF299804 或载体对照,以及 1–5 nM GST-erbB。摇动反应物并在室温下孵育六分钟。除去反应混合物以停止激酶反应后,使用洗涤缓冲液(PBS 中的 0.1% Tween 20)清洁孔。磷酸化酪氨酸残基的检测涉及在含有 3% BSA 和 0.05% Tween 20 的 PBS 中添加 0.2 μg/mL 抗磷酸酪氨酸抗体(Oncogene Ab-4;50 μL/孔)以及稀释的辣根过氧化物酶 (HRP)。然后在室温下摇动25分钟。消除抗体后,使用洗涤缓冲液洗涤板。每孔中加入 50 μL HRP 底物(SureBlue3,3,5,5-四甲基联苯胺,或 TMB),并在室温下摇动孵育 10 至 20 分钟。将 50 μL 终止溶液 (0.09 N H2SO4) 添加到 TMB 反应中以终止反应。 450 nm 处的吸光度用于量化信号。使用中值效应法来确定 PF299804 的 IC50 值。
将重组EGFR(野生型、T790M、L858R/T790M)、HER2和HER4激酶结构域分别与ATP及特异性多肽底物在系列稀释的达可替尼(PF-00299804, PF-299)(0.001-100μM)存在下孵育,反应在37°C下进行60分钟,采用放射免疫法检测磷酸化底物。通过与溶媒对照组的放射性对比计算抑制率,从量效曲线中得出IC₅₀值[1] 为证实不可逆结合特性,将药物与EGFR T790M激酶结构域预孵育30分钟后再加入ATP和底物。加入终止缓冲液终止反应,定量磷酸化水平以验证时间依赖性抑制活性[1] |
| 细胞实验 |
在 24 孔板中,以每孔 5×10 3 至 5×10 4 细胞接种重复细胞,并计算生长抑制数据。简而言之,通过添加 10 μM 达克替尼并在铺板后第二天在 12 个浓度范围内进行 2 倍稀释来生成剂量反应曲线。还接种了不含药物的对照孔。在第一天添加药物时对细胞进行计数,并在实验结束(即六天后)时再次对细胞进行计数。使用 Coulter Z1 粒子计数器,在胰蛋白酶消化后将细胞放入等酮溶液中后立即进行计数。使用 Coulter Vi-Cell 计数器对悬浮培养物进行计数 [2]。
- 对于肺癌细胞系:将具有不同EGFR突变(如激活突变和T790M耐药突变)的肺癌细胞系培养在合适的生长培养基中。将细胞以特定密度接种于96孔板中。过夜孵育使细胞贴壁后,将不同浓度(范围从低纳摩尔到微摩尔水平)的达可替尼加入孔中。在一定的孵育时间(通常为48 - 72小时)后,使用如MTT法或基于ATP的细胞活力测定法测量细胞活力。根据这些测定获得的吸光度或发光值计算细胞增殖的抑制率,并生成剂量 - 反应曲线以确定IC50值 [1] - 对于乳腺癌细胞系:将HER2扩增的乳腺癌细胞系培养在合适的培养基中。将细胞铺在96孔板中。细胞贴壁后,加入不同浓度的达可替尼。随时间监测细胞生长情况,例如,在特定时间点(如24、48和72小时)使用细胞计数器计数细胞数量,或使用如XTT法测量细胞的代谢活性。在达可替尼处理后,还对细胞裂解物进行蛋白质免疫印迹分析。通过在合适的裂解缓冲液中裂解细胞来制备细胞裂解物。蛋白质通过SDS - PAGE电泳分离,然后转移到硝酸纤维素膜上。用针对HER2、磷酸化HER2、AKT、磷酸化AKT、ERK和磷酸化ERK的抗体对膜进行检测,以评估达可替尼对HER2信号通路的影响 [2] 将NCI-H1975、HCC827/T790M、JIMT-1和BT474-HR细胞以5×10³个细胞/孔接种到96孔板中,用达可替尼(PF-00299804, PF-299)(0.01-1μM)处理72小时,采用四唑盐法检测细胞活性并计算IC₅₀值[1,2] 用0.05-0.2μM药物处理细胞24小时,裂解后通过蛋白质印迹法检测磷酸化EGFR/HER2、AKT、ERK1/2、周期蛋白D1、p27、切割型caspase-3、PARP和GAPDH的表达[1,2] 用0.08-0.2μM药物处理JIMT-1细胞24小时,进行细胞周期分析。细胞经70%乙醇固定,碘化丙啶染色后通过流式细胞术分析[2] 用0.03-0.1μM药物处理NCI-H1975细胞14天,进行克隆形成实验,随后固定、染色并计数克隆数[1] |
| 动物实验 |
小鼠:体内研究采用6至8周龄的裸鼠(nu/nu)。每只小鼠右下侧腹部皮下注射5×10⁶个HCC827-GFP或HCC827-Del/T790M肺癌细胞悬液(溶于0.2 mL PBS)。ZD1839治疗组每组接种5只小鼠HCC827-GFP或HCC827-Del/T790M细胞。每周两次使用游标卡尺测量肿瘤大小。肿瘤体积计算公式为:长×宽²×0.5²。每日检查小鼠的体重和一般健康状况。当肿瘤平均体积达到400至500 mm³时,将小鼠随机分为两组,分别接受两种治疗。ZD1839每日口服一次,剂量为150 mg/kg/d。达可替尼每日口服一次,剂量为10 mg/kg/d。当对照组肿瘤的平均体积达到2000 mm³时,实验终止。
- 在肺癌异种移植模型中:将携带EGFR或ERBB2突变的人肺癌细胞系皮下注射到裸鼠侧腹。当肿瘤达到一定体积(通常约为100-200 mm³)后,将小鼠随机分为治疗组和对照组。达可替尼溶于合适的溶剂中(例如DMSO和PEG 400的生理盐水混合物)。治疗组小鼠每日口服一次特定剂量(例如10-50 mg/kg)的药物,持续一定时间(通常为2-4周)。每周使用游标卡尺测量两次肿瘤体积,并监测小鼠的体重。肿瘤体积采用以下公式计算:体积 = 长度 × 宽度² × 0.5。治疗结束后,处死小鼠,切除肿瘤进行进一步分析,例如免疫组织化学染色以评估 EGFR、ERBB2 及其磷酸化形式的表达[1]。 携带 NCI-H1975 异种移植瘤(100-150 mm³)的裸鼠被随机分为对照组和治疗组。达可替尼(PF-00299804,PF-299)悬浮于 0.5% 羧甲基纤维素溶液中,以 15 mg/kg/天的剂量口服给药,持续 21 天。每3天测量一次肿瘤体积,处死小鼠并收集肿瘤组织进行EGFR磷酸化Western blot分析[1]。 携带JIMT-1异种移植瘤的裸鼠以20 mg/kg/天的剂量口服药物,持续28天。每日记录生存时间,并对肿瘤组织进行Ki-67(增殖标志物)免疫组化染色[2]。 为进行剂量反应研究,携带HCC827/T790M异种移植瘤的裸鼠以10、15或25 mg/kg/天的剂量口服达可替尼(PF-00299804,PF-299),持续24天。在治疗结束时测量肿瘤重量以计算抑制率[1]。 |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
单次和多次剂量范围研究表明,达可替尼呈线性药代动力学。食物或服用抗酸剂似乎不影响其吸收和分布。连续4天服用45 mg后,血浆峰浓度为104 ng/ml。报道的AUC0-24h和tmax分别为2213 ng·h/mL和6小时。此外,口服给药后,绝对口服生物利用度为 80%。 给药剂量的 79% 从粪便中排出,其中 20% 为未修饰的达克替尼;3% 从尿液中排出,其中不足 1% 为未修饰形式。 据报道,达克替尼的分布容积为 2415 L。 达克替尼的几何表观清除率为 27.06 L/h。 代谢/代谢物 达克替尼主要通过氧化和结合代谢,其代谢主要受谷胱甘肽和细胞色素 P450 酶的活性影响。代谢后,其主要循环代谢物是 O-去甲基达克替尼,命名为 PF-05199265。该代谢物已被证实主要由 CYP2D6 的氧化步骤生成,CYP2C9 的作用较小。后续代谢步骤主要由 CYP3A4 介导,生成更小的代谢物。这些代谢研究表明,达可替尼能强烈抑制 CYP2D6 的活性。 生物半衰期 据报道,达可替尼的半衰期长达 70 小时。 达可替尼(PF-00299804,PF-299)在小鼠单次口服 15 mg/kg 剂量后的生物利用度约为 80%。血浆半衰期约为 9.5 小时,给药后 1.5 小时达到最大血浆浓度 (Cmax) 为 4.6 μg/mL [1] 在大鼠中,口服 20 mg/kg 后,24 小时 AUC₀-24h 为 58.3 μg·h/mL。该药物广泛分布于肿瘤组织、肝脏和肺脏中,肿瘤与血浆浓度比约为 3.0 [2] |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
在早期的大型临床试验中,达克替尼治疗期间血清转氨酶水平升高较为常见,接受标准剂量治疗的患者中约有 40% 出现这种情况。然而,大多数升高是短暂且无症状的,很少导致剂量调整或停药。仅有 1.4% 的患者出现血清 ALT 升高超过正常值上限 5 倍的情况,这一比例低于其他 EGFR 抑制剂,例如厄洛替尼和吉非替尼。血清碱性磷酸酶升高也有发生,但并不常见。未出现伴有黄疸的临床明显肝损伤病例。然而,达克替尼的临床经验有限。 可能性评分:E(未经证实,但怀疑是临床上明显的肝损伤的罕见原因)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 目前尚无关于达克替尼在哺乳期临床应用的信息。由于达克替尼与血浆蛋白的结合率高达98%,因此其在乳汁中的含量可能很低。然而,由于达可替尼可能对母乳喂养的婴儿产生毒性,且其半衰期为70小时,因此生产商建议在达可替尼治疗期间以及末次给药后至少17天内停止母乳喂养。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白结合 已知达可替尼的蛋白结合率为98%。 小鼠以15 mg/kg/天的剂量接受达可替尼(PF-00299804,PF-299)治疗21天后,体重略有下降(约7%),但未出现明显的肝肾毒性。血清ALT、AST和肌酐水平均在正常范围内[1] 通过平衡透析法测定,该药物在人血浆中的血浆蛋白结合率约为94%。在长期毒性研究(28天,20 mg/kg/天,口服)中,大鼠未出现严重的血液学或胃肠道毒性[2] |
| 参考文献 |
|
| 其他信息 |
达可替尼属于喹唑啉类化合物,其化学名称为7-甲氧基喹唑啉-4,6-二胺,其中4位氨基被3-氯-4-氟苯基取代,6位氨基被(E)-4-(哌啶-1-基)丁-2-烯酰基取代。它是一种表皮生长因子受体拮抗剂和抗肿瘤药物。它属于喹唑啉类、哌啶类、烯酰胺类、单氯苯类、单氟苯类、叔胺类、仲胺类和仲羧酰胺类化合物。
达可替尼(Dacomitinib),设计为(2E)-N-16-4-(哌啶-1-基)丁-2-烯酰胺,是一种口服高选择性喹唑啉酮类化合物,属于第二代酪氨酸激酶抑制剂,其特征在于与表皮生长因子受体家族激酶结构域的ATP结构域发生不可逆结合。达可替尼由辉瑞公司研发,并于2018年9月27日获得美国食品药品监督管理局(FDA)批准。文献中的一些证据表明,达可替尼在卵巢上皮癌模型中具有治疗潜力,但仍需进一步研究。 达可替尼是一种多激酶受体抑制剂,用于治疗携带表皮生长因子受体基因(EGFR)激活突变的非小细胞肺癌。达可替尼治疗期间血清转氨酶短暂升高的发生率较高,但尚未发现与临床上明显的急性肝损伤病例相关。 达可替尼是一种高选择性、口服生物利用度高的小分子HER家族酪氨酸激酶抑制剂,具有潜在的抗肿瘤活性。达可替尼特异性且不可逆地结合并抑制人Her-1、Her-2和Her-4,从而抑制过度表达这些受体的肿瘤细胞的增殖并诱导其凋亡。 药物适应症 达可替尼适用于一线治疗经FDA批准的检测方法验证的表皮生长因子受体(EGFR)19号外显子缺失或21号外显子L858R突变的转移性非小细胞肺癌(NSCLC)患者。肺癌是癌症死亡的主要原因,NSCLC占肺癌病例的85%。在NSCLC病例中,约75%的患者确诊时已处于转移性和晚期,生存率仅为5%。 EGFR 基因突变占非小细胞肺癌 (NSCLC) 病例的 60% 以上,EGFR 过表达与淋巴结转移频繁和化疗敏感性差相关。 FDA 标签 Vizimpro 作为单药疗法,适用于一线治疗携带表皮生长因子受体 (EGFR) 激活突变的局部晚期或转移性非小细胞肺癌 (NSCLC) 成人患者。 作用机制 达可替尼是一种不可逆的小分子抑制剂,可抑制人表皮生长因子受体 (EGFR) 家族(EGFR/HER1、HER2 和 HER4)酪氨酸激酶的活性。它通过与 HER 受体催化结构域中的半胱氨酸残基共价结合而实现不可逆抑制。达可替尼的 IC50 为 6 nmol/L。 ErbB 或表皮生长因子 (EGF) 家族通过激活下游信号转导通路(例如 Ras-Raf-MAPK、PLCγ-PKC-NF-κB 和 PI3K/AKT),在肿瘤生长、转移和治疗耐药中发挥作用。这种激活是通过酪氨酸激酶驱动的羧基末端磷酸化实现的。约 40% 的病例显示 EGFR 基因扩增,50% 的病例存在 EGFRvIII 突变,该突变代表一种缺失,导致受体酪氨酸激酶结构域持续激活。 药效学 临床前数据显示,达克替尼可增强表皮生长因子受体激酶结构域的抑制作用,并增强携带耐药突变(例如 T790M)的细胞系的活性。这种活性进一步显著降低了 EGFR 磷酸化水平和细胞活力。在这些研究中,使用了携带 L858R/T790M 突变的非小细胞淋巴瘤癌细胞系,观察到 IC50 值约为 280 nmol/L。在化疗后进展的晚期非小细胞肺癌患者的临床试验中,客观缓解率为 5%,无进展生存期为 2.8 个月,总生存期为 9.5 个月。此外,I/II 期研究显示,即使酪氨酸激酶抑制剂治疗失败,达可替尼仍具有积极疗效。在携带 EGFR 激活突变的晚期或转移性非小细胞肺癌患者中开展的 III 期临床试验(ARCHER 1050)报告称,与吉非替尼相比,达可替尼显著改善了患者的无进展生存期。 - 达可替尼 是一种不可逆的泛 ERBB 抑制剂。它与 ERBB 家族成员催化结构域中 ATP 结合位点的亲核半胱氨酸残基共价结合,导致其酪氨酸激酶活性不可逆抑制。这种抑制作用阻断了对细胞增殖、存活和迁移至关重要的下游信号级联,使其成为ERBB家族成员发生突变和/或扩增的癌症的潜在治疗药物[1] 达可替尼(PF-00299804,PF-299)是一种不可逆的泛HER酪氨酸激酶抑制剂,它与EGFR、HER2和HER4的ATP结合口袋共价结合,阻断参与肿瘤增殖和存活的下游信号通路[1] 它被开发用于克服非小细胞肺癌和乳腺癌中对第一代EGFR抑制剂(例如ZD1839)和抗HER2疗法(例如曲妥珠单抗)的耐药性。临床前数据支持其进入临床试验[2] 该药物已获得FDA批准,用于一线治疗EGFR 19号外显子缺失或21号外显子L858R突变的转移性非小细胞肺癌[1] |
| 分子式 |
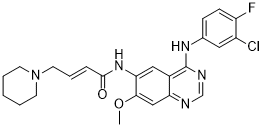
C24H25CLFN5O2
|
|---|---|
| 分子量 |
469.9390
|
| 精确质量 |
469.17
|
| 元素分析 |
C, 61.34; H, 5.36; Cl, 7.54; F, 4.04; N, 14.90; O, 6.81
|
| CAS号 |
1110813-31-4
|
| 相关CAS号 |
Dacomitinib hydrate;1042385-75-0;Dacomitinib-d10 dihydrochloride;Dacomitinib-d10
|
| PubChem CID |
11511120
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
665.7±55.0 °C at 760 mmHg
|
| 熔点 |
184-187 ºC
|
| 闪点 |
356.4±31.5 °C
|
| 蒸汽压 |
0.0±2.0 mmHg at 25°C
|
| 折射率 |
1.663
|
| LogP |
4.4
|
| tPSA |
79.4
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
665
|
| 定义原子立体中心数目 |
0
|
| SMILES |
COC1=C(C=C2C(=C1)N=CN=C2NC3=CC(=C(C=C3)F)Cl)NC(=O)/C=C/CN4CCCCC4
|
| InChi Key |
LVXJQMNHJWSHET-AATRIKPKSA-N
|
| InChi Code |
InChI=1S/C24H25ClFN5O2/c1-33-22-14-20-17(24(28-15-27-20)29-16-7-8-19(26)18(25)12-16)13-21(22)30-23(32)6-5-11-31-9-3-2-4-10-31/h5-8,12-15H,2-4,9-11H2,1H3,(H,30,32)(H,27,28,29)/b6-5+
|
| 化学名 |
(E)-N-[4-(3-chloro-4-fluoroanilino)-7-methoxyquinazolin-6-yl]-4-piperidin-1-ylbut-2-enamide
|
| 别名 |
Vizimpro; PF-00299804; PF00299804; PF 00299804; PF-299; PF299804; PF-299804; PF 299804; PF299; PF 299; dacomitinib
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.32 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (5.32 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.32 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 1% DMSO+30% polyethylene glycol+1% Tween 80, pH 9: 10mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1279 mL | 10.6397 mL | 21.2793 mL | |
| 5 mM | 0.4256 mL | 2.1279 mL | 4.2559 mL | |
| 10 mM | 0.2128 mL | 1.0640 mL | 2.1279 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT04721106
Conditions:Lung NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT03810807
Conditions:Metastatic Non-small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT04504071
Conditions:Non-small Cell Lung Cancer Metastatic|EGF-R Positive Non-Small Cell Lung Cancer
Title:Dacomitinib Plus PD-0325901 in Advanced KRAS Mutant NSCLC
Status:Completed
updateDate:2025-07-08
Ctid:NCT02039336
Link: https://clinicaltrials.gov/ct2/show/NCT02039336
Conditions:Colorectal CancerLink: https://clinicaltrials.gov/ct2/show/NCT04027647
Conditions:NSCLC Stage IIIB|NSCLC Stage IIIC|NSCLC Stage IV|Recurrent NSCLC|EGFR Positive Non-Small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT04946968
Conditions:Advanced Solid Tumours|Non-small Cell Lung Cancer|Head and Neck Squamous Cell CarcinomaLink: https://clinicaltrials.gov/ct2/show/NCT04609319
Conditions:Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT03755102
Conditions:EGFR Gene Mutation|Lung Cancer|Lung Cancer MetastaticLink: https://clinicaltrials.gov/ct2/show/NCT05834348
Conditions:Non-Small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT04423185
Conditions:Rare TumorLink: https://clinicaltrials.gov/ct2/show/NCT00728390
Conditions:Carcinoma, Non-Small Cell|Neoplasm MetastasisLink: https://clinicaltrials.gov/ct2/show/NCT04511533
Conditions:Metastatic Non Small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT03878524
Conditions:Accelerated Phase Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Anatomic Stage IV Breast Cancer AJCC v8|Anemia|Ann Arbor Stage III Hodgkin Lymphoma|Ann Arbor Stage III Non-Hodgkin Lymphoma|Ann Arbor Stage IV Hodgkin Lymphoma|Ann Arbor Stage IV Non-Hodgkin Lymphoma|Atypical Chronic Myeloid Leukemia, BCR-ABL1 Negative|Blast Phase Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Castration-Resistant Prostate Carcinoma|Chronic Phase Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Hematopoietic and Lymphoid System Neoplasm|Locally Advanced Pancreatic Adenocarcinoma|Metastatic Breast Carcinoma|Metastatic Malignant Solid Neoplasm|Metastatic Pancreatic Adenocarcinoma|Myelodysplastic/Myeloproliferative Neoplasm With Ring Sideroblasts and Thrombocytosis|Myelodysplastic/Myeloproliferative Neoplasm, Unclassifiable|Primary Myelofibrosis|Recurrent Acute Lymphoblastic Leukemia|Recurrent Acute Myeloid Leukemia|Recurrent Chronic Lymphocytic Leukemia|Recurrent Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Recurrent Hematologic Malignancy|Recurrent Hodgkin Lymphoma|Recurrent Myelodysplastic Syndrome|Recurrent Myelodysplastic/Myeloproliferative Neoplasm|Recurrent Myeloproliferative Neoplasm|Recurrent Non-Hodgkin Lymphoma|Recurrent Plasma Cell Myeloma|Recurrent Small Lymphocytic Lymphoma|Refractory Acute Lymphoblastic Leukemia|Refractory Acute Myeloid Leukemia|Refractory Chronic Lymphocytic Leukemia|Refractory Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Refractory Chronic Myelomonocytic Leukemia|Refractory Hematologic Malignancy|Refractory Hodgkin Lymphoma|Refractory Malignant Solid Neoplasm|Refractory Myelodysplastic Syndrome|Refractory Myelodysplastic/Myeloproliferative Neoplasm|Refractory Non-Hodgkin Lymphoma|Refractory Plasma Cell Myeloma|Refractory Primary Myelofibrosis|Refractory Small Lymphocytic Lymphoma|Stage II Pancreatic Cancer AJCC v8|Stage III Pancreatic Cancer AJCC v8|Stage IV Pancreatic Cancer AJCC v8|Stage IV Prostate Cancer AJCC v8|Unresectable Pancreatic AdenocarcinomaLink: https://clinicaltrials.gov/ct2/show/NCT02925234
Conditions:Cancer|Tumors|Neoplasm|NeoplasiaLink: https://clinicaltrials.gov/ct2/show/NCT01774721
Conditions:Non-small Cell Lung Cancer With EGFR-Activating MutationsLink: https://clinicaltrials.gov/ct2/show/NCT01000025
Conditions:Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT05271916
Conditions:Non-Small Cell Lung Cancer (NSCLC)Link: https://clinicaltrials.gov/ct2/show/NCT04811001
Conditions:NSCLCLink: https://clinicaltrials.gov/ct2/show/NCT01920061
Conditions:NeoplasmLink: https://clinicaltrials.gov/ct2/show/NCT01520870
Conditions:Glioblastoma|Brain Tumor, RecurrentLink: https://clinicaltrials.gov/ct2/show/NCT01728233
Conditions:Penile Neoplasms|Carcinoma, Squamous CellLink: https://clinicaltrials.gov/ct2/show/NCT04768491
Conditions:EGFR Activating Mutation|NSCLC Stage IV|NSCLC Stage IIIB|NSCLC, RecurrentLink: https://clinicaltrials.gov/ct2/show/NCT00768664
Conditions:Head and Neck NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT04675008
Conditions:Non Small Cell Lung Cancer|Brain MetastasesLink: https://clinicaltrials.gov/ct2/show/NCT03865446
Conditions:Severe Hepatic ImpairmentLink: https://clinicaltrials.gov/ct2/show/NCT04575415
Conditions:NSCLCLink: https://clinicaltrials.gov/ct2/show/NCT00553254
Conditions:Carcinoma, Non Small Cell LungLink: https://clinicaltrials.gov/ct2/show/NCT02382796
Conditions:NSCLCLink: https://clinicaltrials.gov/ct2/show/NCT04339829
Conditions:Dacomitinib for EGFR Mutated Non-Small Cell Lung Cancer (NSCLC) With Brain MetastasesLink: https://clinicaltrials.gov/ct2/show/NCT01441128
Conditions:Carcinoma, Non-Small Cell Lung|Adenocarcinoma|Carcinoma, Squamous Cell|Carcinoma, Large CellLink: https://clinicaltrials.gov/ct2/show/NCT00548093
Conditions:Carcinoma, Non Small Cell LungLink: https://clinicaltrials.gov/ct2/show/NCT01465802
Conditions:Non Small Cell Lung Cancer (NSCLC)Link: https://clinicaltrials.gov/ct2/show/NCT00818441
Conditions:Carcinoma, Non-small CellLink: https://clinicaltrials.gov/ct2/show/NCT00225121
Conditions:NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT01112527
Conditions:Glioblastoma|GBM|Glioblastoma MultiformeLink: https://clinicaltrials.gov/ct2/show/NCT00728468
Conditions:Advanced Malignant Solid TumorsLink: https://clinicaltrials.gov/ct2/show/NCT01858389
Conditions:Non-small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT01360554
Conditions:Non-Small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT02047747
Conditions:Brain CancerLink: https://clinicaltrials.gov/ct2/show/NCT02268747
Conditions:Skin Squamous Cell CancerLink: https://clinicaltrials.gov/ct2/show/NCT01737008
Conditions:Squamous Cell Carcinoma of the Head and NeckLink: https://clinicaltrials.gov/ct2/show/NCT01152853
Conditions:Advanced Gastric Cancer|HER2Link: https://clinicaltrials.gov/ct2/show/NCT01121575
Conditions:Non Small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT01796327
Conditions:Healthy VolunteersLink: https://clinicaltrials.gov/ct2/show/NCT00769067
Conditions:Non-small Cell Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT01484847
Conditions:Head and Neck Squamous Cell CarcinomaLink: https://clinicaltrials.gov/ct2/show/NCT01116843
Conditions:Oral Cavity CancerLink: https://clinicaltrials.gov/ct2/show/NCT02097433
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT01449201
Conditions:Head Neck Cancer Squamous Cell Metastatic|Head Neck Cancer Squamous Cell RecurrentLink: https://clinicaltrials.gov/ct2/show/NCT01571388
Conditions:Healthy|Otherwise Healthy Volunteers With Mild or Moderate Hepatic DysfunctionLink: https://clinicaltrials.gov/ct2/show/NCT01702506
Conditions:Healthy VolunteersLink: https://clinicaltrials.gov/ct2/show/NCT00971191
Conditions:Non-Small Cell Lung Cancer (NSCLC)Link: https://clinicaltrials.gov/ct2/show/NCT00783328
Conditions:NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT00999817
Conditions:Healthy VolunteersLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2012-003590-25
Condition:locally advanced/metastatic squamous cell carcinoma of the penisNeoplasie spinocellulari del pene in fase localmente avanzata/metastaticaLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-004671-37
Condition:Patients with recurrent glioblastomaPacientes con glioblastoma recurrenteLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-022656-22
Condition:Non-small cell lung cancerLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-016509-41
Condition:Incurable stage IIIB/IV non-small cell lung cancer after failure of standard therapy for advanced or metastatic disease.Carcinoma polmonare non a piccole cellule stadio IIIB/IV incurabile dopo fallimento della terapia standard per la malattia avanzata o metastatica.Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-005235-14
Condition:Non-Small cell Lung Cancer
Inhibitory concentration and cell type.
Effects of dacomitinib on cell cycle.Cancer Res.2007 Dec 15;67(24):11924-32. |
The effects of dacomitinib on total and phosphorylated HER2, EGFR, HER4, AKT, and ERK.
Chemical structures of investigated molecules in this article.Cancer Res.2007 Dec 15;67(24):11924-32. |
Effects of dacomitinib on apoptosis.Cancer Res.2007 Dec 15;67(24):11924-32. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
")
")
")
")
")
 463611831
463611831
