| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|

| 靶点 |
DNMT1; DNMT3A; DNMT3B
DNA methyltransferases (DNMTs) (IC₅₀ = ~0.15 μM for recombinant human DNMT1; IC₅₀ = ~0.3 μM for DNMT3a; IC₅₀ = ~0.4 μM for DNMT3b; acts as a mechanism-based inhibitor by incorporating into DNA and covalently trapping DNMTs) [2] - DNMT1 (major target) (EC₅₀ = ~0.2 μM for DNA demethylation in HeLa cells; no significant inhibition of other DNA-modifying enzymes (e.g., DNA polymerase, ligase) with IC₅₀ > 10 μM) [3] |
|---|---|
| 体外研究 (In Vitro) |
接触地西他滨 96 小时后,地西他滨治疗显着降低了 SNU719、NCC24 和 KATOIII 的细胞增殖。地西他滨导致 EBVaGC 中 G2/M 期停滞和死亡,降低侵袭能力,并上调 EBVaGC 中 E-钙粘蛋白表达 [1]。只有高剂量 (10 μM) 地西他滨(0.1-1 μM;24-72 小时)才会产生 G2 期停滞,并伴有 G1 细胞减少 [3]。地西他滨上调 HeLa 细胞中的 DCTPP1 和 dUTPase 表达 [4]。
本研究探讨了DNA去甲基化剂地西他滨对eb病毒相关胃癌(EBVaGC)的作用。地西他滨抑制EBVaGC细胞生长,诱导G2/M阻滞和凋亡。地西他滨处理后,E-cadherin表达上调,细胞运动明显受到抑制。p73和RUNX3启动子区域去甲基化,地西他滨上调其表达。它们增强p21的转录,通过下调c-Myc诱导G2/M阻滞和细胞凋亡。地西他滨还诱导了BZLF1在SNU719中的表达。诱导EBV裂解感染是引起宿主细胞凋亡的另一种方法。本研究首次报道了去甲基化剂在EBVaGC中抑制肿瘤细胞增殖和上调E-cadherin的有效性。 [1] DNA甲基转移酶抑制剂阿扎胞苷和地西他滨是表观遗传癌症治疗的典型药物。为了表征阿扎胞苷和地西他滨的去甲基化活性,我们用这两种药物治疗结肠癌和白血病细胞,并对超过14,000个基因启动子进行了基于阵列的DNA甲基化分析。此外,将药物诱导的去甲基化与缺乏DNA甲基转移酶1 (DNMT1)和DNMT3B的等基因结肠癌细胞的甲基化模式进行了比较。我们发现药物诱导的去甲基化模式具有高度特异性、非随机性和可重复性,表明在复制后特异性位点发生了靶向再甲基化。相应地,我们发现CG岛中的CG二核苷酸优先重新甲基化,表明DNA序列背景的作用。我们还在结肠癌或白血病细胞系中发现了一个从未被药物治疗去甲基化的基因子集。这些去甲基化抗性基因在胚胎干细胞中富集了Polycomb suppression Complex 2组分,并富集了去甲基化基因中不存在的转录因子结合基序。我们的研究结果为阿扎胞苷和地西他滨诱导的DNA甲基化模式提供了详细的见解,并表明药物诱导的DNA去甲基化涉及复杂的调节机制。[3] 1. EB病毒相关胃癌(EBV-GC)抗增殖活性:Decitabine(NSC-127716) 抑制EBV-GC细胞系(SGC-7901/EBV、AGS/EBV)增殖,IC₅₀分别为≈0.5 μM和≈0.7 μM(MTT实验,72 h处理)。1 μM剂量下,SGC-7901/EBV细胞活力降低约65%,AGS/EBV降低约58%,而对正常胃上皮细胞(GES-1)几乎无影响(IC₅₀>5 μM)[1] 2. EBV-GC中E-钙粘蛋白上调:Decitabine(0.2–1 μM处理72 h)剂量依赖性增加SGC-7901/EBV细胞E-钙粘蛋白表达。Western blot显示1 μM时表达增加3.2倍,qRT-PCR显示E-钙粘蛋白mRNA增加2.8倍;ChIP-qPCR证实DNMT1与E-钙粘蛋白启动子的结合减少60%(1 μM时),与DNA去甲基化一致[1] 3. 三阴性乳腺癌(TNBC)敏感性:Decitabine 选择性抑制高DNMT表达的TNBC细胞系(如MDA-MB-231,IC₅₀≈0.3 μM;BT-549,IC₅₀≈0.4 μM),对低DNMT的TNBC细胞(如MCF-10A,IC₅₀>4 μM)疗效较低。0.5 μM时,qRT-PCR显示p16^(INK4a) mRNA增加3.5倍,BRCA1 mRNA增加2.2倍[6] 4. 免疫刺激作用:低剂量Decitabine(0.1 μM处理48 h)诱导多种癌细胞(如B16黑色素瘤、CT26结肠癌)表达CD80。流式细胞术显示B16细胞中CD80⁺细胞比例从≈5%升至≈35%;此效应增强肿瘤特异性细胞毒性T淋巴细胞(CTL)活性:共培养实验中CTL对B16细胞的杀伤率增加约40%[7] 5. DNA掺入与突变效应:Decitabine(0.1–1 μM)可掺入HeLa细胞DNA,0.5 μM时每10,000个核苷酸中约掺入1个(LC-MS/MS检测);使DNA突变率增加约2.5倍(0.5 μM,72 h),但不诱导显著DNA双链断裂(γ-H2AX Western blot:较对照组增加<1.2倍)[8] 6. 核苷酸水解酶调控:Decitabine(0.5 μM)使HCT116细胞中DCTPP1(脱氧胞苷三磷酸焦磷酸酶1)表达增加约1.8倍(qRT-PCR);沉默DCTPP1可增强Decitabine的抗增殖活性(IC₅₀从0.4 μM降至0.15 μM),表明DCTPP1介导细胞耐药[4] |
| 体内研究 (In Vivo) |
在雌性 CD-1 小鼠中,地西他滨(1.0 mg/kg,口服)与四氢尿苷(THU)联合使用会导致严重毒性,并增加与地西他滨血浆水平相关的地西他滨毒性的易感性[5]。已建立 EL4 肿瘤的 C57BL/6 小鼠在给予地西他滨(1.0 mg/kg;腹腔注射;每天一次,持续 5 天)后显示出消退 [7]。
癌细胞缺乏免疫原性被认为是其无法诱导肿瘤特异性T细胞反应的主要原因。在本文中,我们提出证据表明,地西他滨(DAC)是一种DNA甲基化抑制剂,目前用于治疗骨髓增生异常综合征(MDS)、急性髓系白血病(AML)和其他恶性肿瘤,能够在小鼠EL4肿瘤模型中引发抗肿瘤细胞毒性T淋巴细胞(CTL)反应。建立EL4肿瘤的C57BL/6小鼠给予DAC (1.0 mg/kg体重)治疗,每天1次,连用5 d。我们发现DAC治疗导致产生IFN-γ的T淋巴细胞浸润到肿瘤中并引起肿瘤排斥反应。CD8(+) T细胞耗竭,而CD4(+) T细胞不耗竭,肿瘤恢复生长。dac诱导的CTL反应似乎是通过诱导肿瘤细胞上CD80的表达而引起的。表观遗传学证据表明,DAC通过CD80基因启动子中CpG二核苷酸位点的去甲基化诱导CD80在EL4细胞中的表达。此外,我们还发现,短暂的低剂量DAC处理可以诱导多种人类癌细胞中的CD80基因表达。本研究首次证明了表观遗传调控可以诱导肿瘤细胞上一种主要的T细胞共刺激分子的表达,从而克服免疫耐受,诱导有效的抗肿瘤CTL反应。这些结果对设计基于dac的癌症免疫疗法具有重要意义。[7] Decitabine (5-aza-2脱氧胞苷;DAC)联合四氢吡啶(THU)是一种治疗镰状细胞病和β-地中海贫血的潜在口服疗法。在小鼠中进行了一项研究,以评估这种联合治疗的安全性,方法是在DAC前1小时口服DAC和THU,连续2天/周,持续9周,然后恢复28天,以支持其临床试验长达9周的持续时间。四氢吡啶,胞苷脱氨酶的竞争性抑制剂,被用于联合提高DAC的口服生物利用度。剂量为167 mg/kg THU,随后是0,0.2,0.4或1.0 mg/kg DAC;THU载药后加入1.0 mg/kg DAC;或者车辆本身。评估的终点是临床观察、体重、食物消耗、临床病理、大体/组织病理、骨髓微核和毒性动力学。在体重、食物消耗、血清化学或尿液分析参数方面没有发现与治疗相关的影响。血浆DAC水平的剂量和性别依赖性变化在1小时内达到Cmax。在测试的1mg /kg剂量下,与单独使用DAC相比,THU增加了DAC的血浆浓度(~ 10倍)。接受高剂量1 mg/kg DAC + THU的女性出现严重毒性,需要在第5周停止治疗。显微镜检查结果的严重程度和发生率呈剂量依赖性增加;结果包括骨髓细胞减少(伴随相应的血液学改变,白细胞、红细胞、血红蛋白、红细胞压积、网状细胞、中性粒细胞和淋巴细胞减少)、胸腺/淋巴细胞减少、肠上皮细胞凋亡和睾丸变性。骨髓微核分析证实骨髓细胞毒性,抑制红细胞生成和遗传毒性。在恢复期之后,观察到这些影响完全或有缓解的趋势。总之,联合治疗导致DAC毒性敏感性增加,与DAC血浆水平相关,女性比男性更敏感。[5] 1. EBV-GC异种移植瘤生长抑制:在荷SGC-7901/EBV皮下异种移植瘤的裸鼠中,Decitabine(0.5 mg/kg,腹腔注射,隔日1次,持续21天)显著抑制肿瘤生长。第21天肿瘤体积:处理组约220 mm³ vs 对照组约780 mm³,肿瘤生长抑制率(TGI)≈72%;肿瘤组织中E-钙粘蛋白蛋白增加2.5倍(Western blot),DNMT1活性降低约60%[1] 2. TNBC异种移植瘤疗效:在荷MDA-MB-231(高DNMT1)异种移植瘤的NOD/SCID小鼠中,Decitabine(0.3 mg/kg,静脉注射,每日1次,持续14天)使肿瘤重量减少约65%(0.18 g vs 对照组0.52 g);肿瘤p16^(INK4a) mRNA增加3.0倍,全局DNA甲基化(5-mC)降低约45%(ELISA)[6] 3. 黑色素瘤免疫治疗效应:在荷B16黑色素瘤异种移植瘤的C57BL/6小鼠中,低剂量Decitabine(0.1 mg/kg,腹腔注射,每3天1次,持续15天)使瘤内CD8⁺ T细胞浸润增加约2.8倍(流式细胞术),肺转移结节减少约55%(HE染色);小鼠生存期从对照组约21天延长至约32天[7] 4. 与四氢尿苷(THU)联用的毒性:CD-1小鼠给予Decitabine(0.2 mg/kg口服)+ THU(2 mg/kg口服,每日1次,持续28天),无显著体重下降(较对照组变化<5%);血清ALT/AST和肌酐水平正常,但观察到轻度骨髓抑制(第14天外周白细胞计数减少约15%,第28天恢复)[5] |
| 酶活实验 |
掺入测定[8]
处理前24小时,将细胞一式三份接种在12孔板中,密度为每孔2×105个细胞,然后与100nM[3H]-地西他滨(或其他浓度,如有指示)一起孵育。孵育24小时(或其他时间段,如果需要)后,用磷酸盐缓冲盐水(PBS)洗涤细胞。对于掺入测量,分别使用DNeasy血液和组织试剂盒或RNeasy试剂盒纯化DNA或RNA,并通过紫外线(UV)光度计进行定量。将纯化的样品与液体闪烁混合物混合,并通过液体闪烁计数测量其放射性。将测量值标准化为DNA(或RNA)的量。如前所述,地西他滨对胞嘧啶的取代百分比计算为作为总胞嘧啶的一部分引入的地西他宾的量。对于竞争实验,细胞要么用100nM[3H]-地西他滨加上增加浓度(100nM,500nM,1μM,2μM)的[14C]-脱氧胞苷处理,要么用100nm[14C]--脱氧胞苷加上增加的[3H]--地西他宾浓度(100nm,500nM,1μM,2μM)处理。24小时后,用PBS洗涤细胞,使用DNeasy血液和组织试剂盒提取DNA,并使用液体闪烁计数进行测量。 DNA甲基化分析[8] 使用DNeasy血液和组织试剂盒从细胞中分离基因组DNA。如前所述,通过毛细管电泳测定全局DNA甲基化水平。 地西他滨(5-aza-2'-脱氧胞苷,aza-dCyd)是一种抗癌药物,临床上用于治疗骨髓增生异常综合征和急性髓性白血病,可以作为一种剂量依赖性的dna去甲基化或基因毒性药物。另一方面,DCTPP1 (dCTP焦磷酸酶1)和dUTPase是两种参与消除非典型核苷酸的“大扫除”核苷酸水解酶。在本研究中,我们发现,暴露于地西他滨的HeLa细胞上调了几种嘧啶代谢酶的表达,包括DCTPP1、dUTPase、dCMP脱氨酶和胸苷酸合成酶,从而表明它们参与了细胞对这种抗癌核苷的反应。我们提出了几条证据支持,除了形成aza-dCTP (5-aza-2'-脱氧胞苷-5'-三磷酸)外,地西他滨的另一种细胞毒性机制可能涉及形成aza-dUMP,一种潜在的胸苷酸合成酶抑制剂。事实上,dUTPase或DCTPP1的下调增强了地西他滨的细胞毒性作用,产生了含有尿嘧啶的三磷酸核苷的积累,以及尿嘧啶在基因组DNA中的错误掺入和双链断裂。此外,DCTPP1以与其天然底物dCTP相似的动力学效率水解地西他滨的三磷酸形式,并阻止地西他滨诱导的整体DNA去甲基化。这些数据表明,核苷酸水解酶DCTPP1和dUTPase是参与地西他滨作用模式的因素,作为改善地西他滨化疗的酶靶点具有潜在价值。[3] 1. DNMT活性抑制实验:将重组人DNMT1(10 nM)、DNMT3a(15 nM)或DNMT3b(15 nM)与生物素化DNA底物(2 μg)、S-腺苷-L-甲硫氨酸(SAM,10 μM)及系列浓度Decitabine(0.01–1 μM)在反应缓冲液(50 mM Tris-HCl pH 7.5、10 mM MgCl₂、1 mM DTT)中37°C孵育2 h;用0.5 M EDTA终止反应,链霉亲和素包被板捕获甲基化DNA,抗5-甲基胞嘧啶(5-mC)抗体检测;测量荧光强度,非线性回归计算IC₅₀[2] 2. DCTPP1活性实验:将重组人DCTPP1(5 nM)与Decitabine三磷酸(dCTP类似物,10 μM)及系列浓度Decitabine(0.1–5 μM)在缓冲液(20 mM HEPES pH 7.4、5 mM MgCl₂)中37°C孵育1 h;焦磷酸检测试剂盒定量释放的焦磷酸,DCTPP1活性以相对于对照组的焦磷酸生成百分比表示[4] 3. DNA掺入实验:HeLa细胞用Decitabine(0.1–1 μM)处理72 h,提取基因组DNA,超声破碎并消化为单核苷酸;通过LC-MS/MS(检测波长260 nm),将Decitabine-单磷酸的峰面积与标准曲线比较,定量掺入量[8] |
| 细胞实验 |
细胞周期分析[1]
细胞类型: HCT116 细胞 测试浓度: 0.1、1、10 µM 孵育时间: 24、48、72 小时 实验结果: 只有高药物浓度 (10 µM) 才会导致 G2 期停滞,同时伴随着 G1 期细胞的减少阶段。 转运试验[8] 如前所述进行运输分析。治疗前24小时,细胞三次接种于12孔板,每孔密度为2 × 105个细胞,然后用100 nM [3H]-地西他滨孵育。经过指定时间的孵育后,用PBS洗涤细胞,用0.2%十二烷基硫酸钠(SDS)裂解。分离样品与液体闪烁鸡尾酒混合,用闪烁计数法测定放射性。与此同时,用比辛胆酸(BCA)蛋白质测定法测量蛋白质浓度,使细胞总摄取与总蛋白质浓度正常化。 细胞周期分析[8] 100 nM 地西他滨处理约1 × 106个细胞。在指定的时间点收集细胞并用冷冻无水乙醇固定。固定后,用PBS洗涤细胞,离心,在染色液(0.1% Triton X-100, 0.2 mg/ml RNase A和20 μg/ml碘化丙啶)中重悬15分钟,37℃,黑暗。流式细胞术分析使用FACS Canto II (BD Biosciences)检测了1万个细胞,并使用FlowJo软件分析数据。 全基因组测序[8] 实验前24 h播种细胞。细胞用100 nM 地西他滨处理24 h,使用dnasy Blood and Tissue kit提取基因组DNA。将DNA剪切成约300 bp的片段。然后连接转接头,选择片段大小并纯化。在Illumina cBot上进行聚类生成。从8个样本(4个对照,4个处理)中生成的聚类在Illumina HiSeq 2000平台上使用101 bp的配对末端reads在一个通道上同时测序。使用FastQC对生成的序列进行质量控制。使用Bowtie2和hg19作为参考完成映射。采用SAMtools进行点突变分析。基因组重排的数量是通过确定不一致对齐的读对的比例来估计的。所有实验均采用独立的生物重复。映射效率和覆盖范围见补充表S2。测序数据已存入SRA数据库,登录号为SRP040672。 1. MTT抗增殖实验(EBV-GC/TNBC):EBV-GC细胞(SGC-7901/EBV、AGS/EBV)或TNBC细胞(MDA-MB-231、BT-549)以3×10³个/孔接种96孔板,过夜培养;加入系列浓度Decitabine(0.01–10 μM),37°C、5% CO₂孵育72 h;每孔加MTT试剂(5 mg/mL,10 μL)孵育4 h,加二甲亚砜(100 μL/孔)溶解甲臜,检测570 nm吸光度,计算IC₅₀[1][6] 2. E-钙粘蛋白表达(Western blot/qRT-PCR):SGC-7901/EBV细胞用Decitabine(0.2–1 μM)处理72 h,提取总蛋白进行Western blot(抗E-钙粘蛋白、抗GAPDH);TRIzol提取总RNA进行qRT-PCR(E-钙粘蛋白、GAPDH引物),相对于对照组定量条带强度和mRNA水平[1] 3. DNMT1结合ChIP-qPCR实验:1 μM Decitabine处理72 h的SGC-7901/EBV细胞用1%甲醛交联,超声破碎染色质,与抗DNMT1抗体或IgG(对照)4°C孵育过夜;蛋白A/G珠捕获免疫复合物,纯化DNA后,用E-钙粘蛋白启动子特异性引物进行qPCR[1] 4. CD80检测(流式细胞术):B16细胞用Decitabine(0.1 μM)处理48 h后收集,用抗CD80-PE抗体染色(30分钟,4°C,避光);流式细胞术分析,计算CD80⁺细胞比例[7] 5. 克隆形成实验(TNBC):MDA-MB-231细胞以200个/孔接种6孔板,贴壁过夜后加入Decitabine(0.1–0.5 μM),每3天换液,培养14天;4%甲醛固定,0.1%结晶紫染色,计数克隆;0.5 μM时克隆形成率降低约80%[6] |
| 动物实验 |
动物/疾病模型: C57BL/6 小鼠(携带 EL4 细胞)[6]
剂量: 1.0 mg/kg 给药途径: 腹腔注射 (ip);每日一次,连续 5 天 实验结果: 即使在停止地西他滨治疗后,仍能持续抑制肿瘤生长。 实验设计 [5] 小鼠被分为四个剂量组和一个溶剂对照组,如表 1 所示。在给予 THU 或其溶剂 1 小时 ± 5 分钟后,以 10 mL/kg 的剂量体积灌胃给予 地西他滨(5-氮杂-2'-脱氧胞苷;DAC) 或其溶剂。 DAC剂量的选择基于一项剂量范围探索研究,该研究中小鼠耐受了6次口服剂量(每周2次),剂量分别为0.1、0.2和0.4 mg/kg DAC,并联合固定剂量167 mg/kg THU。固定剂量THU(500 mg/m²)和THU与DAC给药之间的最佳时间间隔(60分钟)的选择基于之前的研究11。小鼠每体表面积剂量转换为每公斤体重剂量是基于美国食品药品监督管理局(FDA)发布的指南中小鼠的米氏常数(km)值。简而言之,小鼠每体表面积剂量(500 mg/m²)除以km值3,即可转换为每公斤体重剂量(167 mg/kg)。该指南中小鼠的工作体重范围为11-34克。本研究中使用的小鼠体重范围为 24-38 克。 毒代动力学[5] 在每次采集样本前,向样本采集管中加入 10 μL/管 10 mg/mL 的 THU 溶液。在研究第 1 天(第 2 至 5 组)和第 58 天(第 2 至 5 组,第 4 组雌性除外),从未禁食、麻醉的毒代动力学动物中通过心脏穿刺采集血样(约 0.5 mL),分别在给予地西他滨(5-氮杂-2'-脱氧胞苷;DAC)后 15、30、60、90、120 和 180 分钟采集。每个时间点,每组每性别 3 只动物。由于第4组雌性动物死亡,在研究第38天对前5只存活的动物进行尸检,并在DAC给药后15、30和120分钟分别从每个时间点采集3只雌性动物的血液样本。所有样本均在目标时间点后5分钟内采集。 1. EBV-GC皮下异种移植模型:将5×10⁶个SGC-7901/EBV细胞(PBS:Matrigel = 1:1)皮下注射到裸鼠(6-8周龄,每组n=6)右侧腹部。当肿瘤体积达到100-150 mm³时,将小鼠随机分为载体组(0.9%生理盐水)和地西他滨组。地西他滨(0.5 mg/kg)每隔一天腹腔注射一次,持续21天。每3天测量一次肿瘤体积(长×宽²/2)和体重。处死小鼠后收集肿瘤组织,用于Western blot和DNMT活性检测[1] 2. TNBC异种移植模型:将2×10⁶个MDA-MB-231细胞(PBS:Matrigel = 1:1)皮下注射到NOD/SCID小鼠(n=6/组)中。当肿瘤体积达到80–100 mm³时,每天静脉注射一次地西他滨(0.3 mg/kg),持续14天。处死小鼠后测量肿瘤重量,并分析肿瘤组织中p16^(INK4a) mRNA和整体甲基化水平[6] 3. 黑色素瘤免疫治疗模型:将1×10⁶个B16细胞皮下注射到C57BL/6小鼠(n=8/组)中。当肿瘤体积达到 50 mm³ 时,每 3 天腹腔注射一次地西他滨(0.1 mg/kg),持续 15 天。采用流式细胞术分析肿瘤内 CD8⁺ T 细胞,并进行 HE 染色后计数肺转移灶。记录生存时间 [7] 4. 亚慢性口服毒性模型(含 THU):将 CD-1 小鼠(每组 n=10)随机分为三组:载体组(0.5% CMC-Na)、地西他滨单药组(0.2 mg/kg 口服)和地西他滨 + THU 联合组(0.2 mg/kg + 2 mg/kg 口服)。每日给药一次,持续 28 天。每周测量体重。在第 14 天和第 28 天检测血清生化指标(ALT、AST、肌酐)和外周血细胞计数 [5] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
地西他滨以 15 mg/m2 的剂量静脉注射,每 8 小时一次,持续 3 天,结果显示 Cmax 为 73.8 ng/mL(变异系数 CV 为 66%),AUC0-∞ 为 163 ngh/mL(CV 为 62%),累积 AUC 为 1332 ngh/mL(95% CI 为 1010-1730)。同样,地西他滨以 20 mg/m² 的剂量,每日一次,每次一小时,连续五天给药,其 Cmax 为 147 ng/mL(变异系数 49%),AUC0-∞ 为 115 ngh/mL(变异系数 43%),累积 AUC 为 570 ngh/mL(95% CI 470-700)。 给药的地西他滨不足 1% 经尿液排泄。 地西他滨的表观分布容积为 4.59 ± 1.42 L/kg。 地西他滨以 15 mg/m² 的剂量,每八小时静脉注射三小时,连续三天给药,其清除率为 125 L/hr/m²(变异系数 53%),而以 210 L/hr/m² 的剂量,其清除率为 210 L/hr/m²(95% CI 47%)。 CV) 20 mg/m²,每日一次,持续 1 小时,连续 5 天。 代谢/代谢物 地西他滨在细胞内经脱氧胞苷激酶、核苷单磷酸激酶和核苷二磷酸激酶的顺序作用磷酸化,然后被 DNA 聚合酶掺入新合成的 DNA 中。未掺入细胞DNA的地西他滨经胞苷脱氨酶脱氨后进一步降解,最终排出体外。 生物半衰期 地西他滨以15 mg/m²的剂量静脉注射,每8小时一次,持续3天,半衰期为0.62小时(变异系数49%);以20 mg/m²的剂量每日一次,持续1小时,持续5天,半衰期为0.54小时(变异系数43%)。 1. 口服生物利用度(与THU联用):CD-1小鼠口服地西他滨(0.2 mg/kg)+ THU(2 mg/kg),口服生物利用度约为40%,而单独使用地西他滨的口服生物利用度约为5%(通过口服与静脉注射0.1 mg/kg的AUC₀₋∞计算)。 THU抑制胞苷脱氨酶,减少地西他滨的降解[5] 2. 血浆药代动力学:在小鼠中,静脉注射地西他滨(0.1 mg/kg)显示Cₘₐₓ = ~0.8 μM,Tₘₐₓ = 0.25 h,t₁/₂ = ~1.2 h,AUC₀₋₂₄ₕ = ~1.5 μM·h。口服地西他滨 + THU(0.2 mg/kg + 2 mg/kg)的 Cₘₐₓ = ~0.5 μM,Tₘₐₓ = ~1 h,t₁/₂ = ~1.8 h [5] 3. 组织分布:用地西他滨(0.5 mg/kg 腹腔注射)给携带 SGC-7901/EBV 异种移植瘤的裸鼠注射。给药后 1 小时,组织浓度(LC-MS/MS)分别为:肿瘤 ~0.6 μM,肝脏 ~1.2 μM,脾脏 ~0.9 μM,肾脏 ~0.7 μM,脑组织 <0.1 μM(未穿透血脑屏障)[1] 4. 排泄:大鼠静脉注射地西他滨(0.3 mg/kg)后,24 小时内约 60% 的药物经尿液排出(30% 为原药,30% 为无活性代谢物:尿嘧啶类似物)。粪便排泄约占 15% [5] |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
在早期使用高剂量地西他滨的临床试验中,高达 16% 的合并肝病或肝转移患者出现血清酶升高,但在无肝病患者中很少见。在随后的研究中,5% 至 15% 的治疗患者报告出现血清 ALT 升高,但所有升高均为自限性,且未报告临床上明显的肝损伤。近期研究报告 7% 至 12% 的治疗患者出现血清胆红素水平升高,但升高迅速消退,且未伴有其他临床或实验室肝损伤证据。建议仅在合并肝病的患者中监测治疗期间的血清酶水平。 口服地西他滨联合西达唑啶的治疗方案与单独静脉注射地西他滨的治疗方案相比,不良事件的发生率和模式似乎相似。在多项前瞻性临床试验中,单周期口服与静脉注射地西他滨引起的转氨酶升高率相似;长期、多疗程的口服固定剂量复方制剂治疗导致20%至37%的患者出现血清转氨酶升高,其中2%至3%的患者转氨酶水平超过正常值上限(ULN)的5倍,但未发现任何可归因于化疗药物的临床明显肝损伤病例。 因此,尽管地西他滨被广泛用于治疗骨髓增生异常综合征(MDS),但尚未有确凿证据表明其与临床明显肝损伤病例相关。然而,由于治疗期间血清酶升高发生率较高,因此很难断言地西他滨完全没有引起药物性肝损伤的风险。 可能性评分:E(未经证实但怀疑,罕见,可导致临床上明显的肝损伤)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 大多数资料认为,母亲接受抗肿瘤药物治疗期间应避免哺乳。间歇性地西他滨治疗期间,如果哺乳期适当,则可能可以安全哺乳;生产商建议在末次给药后停止哺乳1周。化疗可能会对母乳的正常微生物群和化学成分产生不利影响。孕期接受化疗的女性更容易出现哺乳困难。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 一项电话随访研究对74名在孕中期或孕晚期于同一中心接受癌症化疗的女性进行了调查,以确定她们产后是否成功进行母乳喂养。仅有34%的女性能够纯母乳喂养婴儿,66%的女性报告存在母乳喂养困难。相比之下,22名在孕期确诊但未接受化疗的母亲的母乳喂养成功率为91%。其他具有统计学意义的相关性包括:1)出现母乳喂养困难的母亲平均接受了5.5个疗程的化疗,而没有出现困难的母亲平均接受了3.8个疗程的化疗; 2)有哺乳困难的母亲平均提前3.4周接受了第一个化疗周期。在接受含氟尿嘧啶方案的9名女性中,8名有哺乳困难。 蛋白结合 地西他滨的血浆蛋白结合率可忽略不计(< 1%)。 1. 急性毒性:在小鼠中,地西他滨的静脉注射LD₅₀约为5 mg/kg;口服LD₅₀>10 mg/kg(由于生物利用度低)。静脉注射 2 mg/kg 时,小鼠出现短暂嗜睡,但在 24 小时内恢复 [5] 2. 亚慢性毒性(与 THU 一起):用地西他滨 + THU(0.2 mg/kg + 2 mg/kg 口服,28 天)治疗的 CD-1 小鼠没有明显的肝/肾损伤(ALT/AST < 1.2 倍 vs. 载体;肌酐正常)。轻度骨髓抑制(第14天中性粒细胞计数减少约15%)是可逆的[5] 3. 癌症模型中的骨髓抑制:用地西他滨(0.5 mg/kg,腹腔注射,21天)治疗的裸鼠在第14天外周血小板减少约20%,到第21天恢复正常。未观察到严重贫血或白细胞减少症[1] 4. 血浆蛋白结合:将地西他滨(1 μM)与人血浆在37°C下孵育1小时。通过超滤(30 kDa截留分子量)分离未结合的药物,并用LC-MS/MS进行测定。血浆蛋白结合率约为15%(低结合)[5] 5. DNA损伤相关毒性:用地西他滨(1 μM,72 h)处理的HeLa细胞中γ-H2AX(DNA双链断裂标志物,Western blot:与载体相比<1.2倍)未见显著增加,表明在治疗浓度下基因毒性较低[8] |
| 参考文献 |
[1]. Decitabine inhibits tumor cell proliferation and up-regulates E-cadherin expression in Epstein-Barr virus-associated gastric cancer. J Med Virol. 2017 Mar;89(3):508-517.
[2]. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. 2009 Jul;109(7):2880-93. [3]. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS One. 2011 Mar 7;6(3):e17388. [4]. The nucleotidohydrolases DCTPP1 and dUTPase are involved in the cellular response to decitabine. Biochem J. 2016 Sep 1;473(17):2635-43. [5]. Subchronic oral toxicity study of decitabine in combination with tetrahydrouridine in CD-1 mice. Int J Toxicol. 2014 Mar-Apr;33(2):75-85. [6]. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest. 2018 Jun 1;128(6):2376-2388. [7]. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumorspecific cytotoxic T lymphocyte responses. PLoS One. 2013 May 9;8(5):e62924. [8]. Quantitative determination of decitabine incorporation into DNA and its effect on mutation rates in human cancer cells. Nucleic Acids Res. 2014 Oct 29; 42(19): e152. |
| 其他信息 |
药效学
地西他滨是天然核苷酸2'-脱氧胞苷的前药类似物,在细胞内磷酸化后可掺入DNA,并对基因表达产生多种影响。使用地西他滨与中性粒细胞减少症和血小板减少症相关。此外,地西他滨可对孕妇的胎儿造成损害;建议在接受地西他滨治疗期间采取有效的避孕措施并避免怀孕。 1. 作用机制:地西他滨是一种核苷类似物,在细胞内磷酸化为活性三磷酸形式(dAza-TP)。它在DNA复制过程中掺入DNA,与DNMTs(尤其是DNMT1)共价结合,并降低DNMT活性。这可诱导全局性和基因特异性DNA去甲基化,重新激活沉默的抑癌基因(例如E-钙黏蛋白、p16^(INK4a)),并抑制癌细胞增殖[2][3][6] 2. 治疗背景:地西他滨已获批用于治疗老年患者的骨髓增生异常综合征(MDS)和急性髓系白血病(AML)。临床前研究支持其在EBV相关胃癌(通过上调E-钙黏蛋白)和三阴性乳腺癌(在DNMT高表达亚型中)中的潜力[1][6] 3. 联合用药策略:与THU联合用药可通过抑制胞苷脱氨酶(降解地西他滨)来提高地西他滨的口服生物利用度。这使得口服给药成为可能,减少了肠外给药的需求[5] 4. 免疫调节作用:低剂量地西他滨可上调癌细胞上的CD80(一种共刺激分子),增强CTL介导的抗肿瘤免疫。这使其成为免疫检查点抑制剂(例如抗PD-1)的潜在联合用药伙伴[7] 5. 耐药机制:DCTPP1(一种核苷酸水解酶)可降解地西他滨的活性三磷酸形式,从而导致细胞耐药。沉默DCTPP1或抑制其活性可使癌细胞对地西他滨敏感[4] |
| 分子式 |
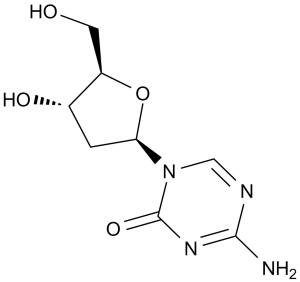
C8H12N4O4
|
|
|---|---|---|
| 分子量 |
228.21
|
|
| 精确质量 |
228.085
|
|
| 元素分析 |
C, 42.10; H, 5.30; N, 24.55; O, 28.04
|
|
| CAS号 |
2353-33-5
|
|
| 相关CAS号 |
|
|
| PubChem CID |
451668
|
|
| 外观&性状 |
White to off-white solid
|
|
| 密度 |
1.9±0.1 g/cm3
|
|
| 沸点 |
485.8±55.0 °C at 760 mmHg
|
|
| 熔点 |
~200 °C (dec.)
|
|
| 闪点 |
247.6±31.5 °C
|
|
| 蒸汽压 |
0.0±2.8 mmHg at 25°C
|
|
| 折射率 |
1.780
|
|
| LogP |
-1.93
|
|
| tPSA |
123.49
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
16
|
|
| 分子复杂度/Complexity |
356
|
|
| 定义原子立体中心数目 |
3
|
|
| SMILES |
O1[C@]([H])(C([H])([H])[C@@]([H])([C@@]1([H])C([H])([H])O[H])O[H])N1C([H])=NC(N([H])[H])=NC1=O
|
|
| InChi Key |
XAUDJQYHKZQPEU-KVQBGUIXSA-N
|
|
| InChi Code |
InChI=1S/C8H12N4O4/c9-7-10-3-12(8(15)11-7)6-1-4(14)5(2-13)16-6/h3-6,13-14H,1-2H2,(H2,9,11,15)/t4-,5+,6+/m0/s1
|
|
| 化学名 |
4-amino-1-((2S,4S,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1,3,5-triazin-2(1H)-one
|
|
| 别名 |
5-Aza-2'-deoxycytidine; deoxyazacytidine; 2353-33-5; Dacogen; 2'-Deoxy-5-azacytidine; 5-Azadeoxycytidine; AzadC; 5-aza-CdR; 5-aza-dCyd; Deoxycytidine; NSC127716; NSC 127716; NSC-127716; dezocitidine; Brand name: Dacogen. Abbreviations: 5AZA; DAC
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (10.95 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (10.95 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (10.95 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 30% propylene glycol, 5% Tween 80, 65% D5W:30mg/mL 配方 5 中的溶解度: 10 mg/mL (43.82 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.3819 mL | 21.9096 mL | 43.8193 mL | |
| 5 mM | 0.8764 mL | 4.3819 mL | 8.7639 mL | |
| 10 mM | 0.4382 mL | 2.1910 mL | 4.3819 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT02316028 | Completed | Drug: Decitabine | Liver Metastasis Colorectal Cancer |
Universitair Ziekenhuis Brussel | March 2014 | Phase 1 Phase 2 |
| NCT05960773 | Recruiting | Drug: Decitabine/cedazuridine | Mesothelioma Malignant Mesothelioma (MM) |
National Cancer Institute (NCI) | January 31, 2024 | Phase 2 |
| NCT05816356 | Recruiting | Drug: Decitabine Drug: Tetrahydrouridine |
Healthy | EpiDestiny, Inc. | March 24, 2023 | Phase 1 |
| NCT04582604 | Recruiting | Drug: modified By/Cy conditioning regimen intensified by Ruxolitinib and Decitabine |
Peripheral Blood Stem Cell Transplantation |
Chinese PLA General Hospital | September 1, 2020 | Phase 1 Phase 2 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
