| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
RIPK3
GSK-843 (3-10 μM; 18 h) induces apoptosis[1]. GSK-843 (0.3-3 μM; 18 h) inhibits virus- and TNF-induced cell necrosis[2]. Researchers previously identified the RIP3i GSK’843 and GSK’872 by screening conventional small-molecule libraries (Kaiser et al., 2013a) (Figure 1A). These compounds bound RIP3 kinase domain with high affinity (IC50 = 8.6 nM and 1.8 nM, respectively; Figure 1B) and inhibited kinase activity (IC50 = 6.5 nM and 1.3 nM, respectively; Figure 1C). When assayed individually at 1 μM, the three structurally distinct compounds failed to inhibit most of 300 human protein kinases tested, with GSK’840 showing the best profile (Figure S1B; Table S1). All compounds failed to inhibit RIP1 kinase when tested directly (data not shown). Taken together, this demonstrates that GSK’840, GSK’843, and GSK’872 bind to RIP3 kinase domain and inhibit enzyme activity with minimal cross-reactivity[1]. |
|---|---|
| 体外研究 (In Vitro) |
GSK-843(3-10 μM;18 小时)诱导细胞凋亡[1]。 GSK-843(0.3-3 μM;18 小时)抑制病毒和 TNF 诱导的细胞坏死[2]。
研究人员之前通过筛选常规小分子文库确定了RIP3i GSK ' 843和GSK ' 872 (Kaiser et al., 2013a)(图1A)。这些化合物以高亲和力结合RIP3激酶结构域(IC50分别为8.6 nM和1.8 nM);图1B)和抑制的激酶活性(IC50分别为6.5 nM和1.3 nM;图1 c)。当在1 μM下单独检测时,这三种结构不同的化合物对300种人蛋白激酶的大部分都没有抑制作用,其中GSK 840表现出最好的抑制效果(图S1B;表S1)。当直接测试时,所有化合物都不能抑制RIP1激酶(数据未显示)。综上所述,这表明GSK ' 840、GSK ' 843和GSK ' 872结合到RIP3激酶结构域,并以最小的交叉反应性抑制酶活性[1]。 GSK'843 以浓度依赖性方式抑制人 HT-29 细胞中 TNF 诱导的坏死性凋亡,其 IC50 比无细胞生化实验高 100 至 1000 倍 [1]。 GSK'843 在 0.04 至 1 μM 的浓度范围内,可抑制小鼠细胞中的坏死性凋亡,这些细胞包括骨髓来源巨噬细胞、硫代乙醇酸盐诱导的腹膜巨噬细胞和 3T3SA 成纤维细胞 [1]。 GSK'843 可抑制由 poly(I:C) 在泛 caspase 抑制剂 zVAD 存在下触发的 Toll 样受体 3 诱导的坏死性凋亡 [1]。 在高浓度(3 μM 和 10 μM)下,GSK'843 会在多种细胞类型中触发 caspase 依赖性凋亡性细胞死亡,该死亡可被 zVAD 逆转 [1]。 这种凋亡的特征包括 caspase-3 被切割、效应 caspase 活性增加、膜起泡以及电镜下的凋亡形态 [1]。 GSK'843 的促凋亡活性需要 RIP3 的存在,这在 RIP3 低表达的 NIH3T3 细胞敏感性降低、Rip3/- MEF 细胞抵抗以及用人类 RIP3 重建的 Rip3/- MEF 恢复敏感性等实验中得到证实 [1]。 GSK'843 诱导的凋亡不依赖于促坏死机器,但依赖于 RHIM 信号传导,因为 RHIM 突变的人类 RIP3 无法赋予敏感性 [1]。 用 GSK'843 处理会以浓度依赖性方式驱动 RIP1-FADD-cFLIPL-Casp8 复合物的组装,尤其是在 zVAD 存在下该复合物更稳定 [1]。 这种凋亡需要 RIP1、FADD、cFLIPL 和 Casp8,但不需要 RIP1 激酶活性,因为来自 Rip1K45A/K45A 激酶失活敲入小鼠的细胞仍然敏感 [1]。 来自 MCMV 的病毒 RIP 激活抑制剂(vIRA)可抑制 GSK'843 诱导的凋亡 [1]。 GSK'843 显示出最小的交叉反应性,在 1 μM 浓度下对测试的 300 种人类蛋白激酶中的大多数没有抑制作用,并且不直接抑制 RIP1 激酶 [1]。 |
| 体内研究 (In Vivo) |
Rip3K51A/K51A激酶死亡敲入小鼠存活[1]
RIP3激酶死亡突变体的行为支持在D161N突变敲入小鼠中观察到的惊人的妊娠中期致命性(Newton等人,2014),并预测无毒突变体将出现相反的结果。当产生Rip3K51A/K51A激酶死亡敲入小鼠时,小鼠明显具有活力和生育能力(图7A和7B)。该突变株对妊娠中期或围产期死亡没有表现出任何易感性。为了确定存活的Rip3K51A/K51A突变体是否像致死性Rip3D161N/D161N突变体(Newton et al., 2014)一样,挽救了Casp8−/−胚胎的胚胎致死性,我们进行了杂交,并以预期的孟德尔频率挽救了存活和可育的Casp8−/−Rip3K51A/K51A小鼠(图7B和S6A)。这扩展了先前对Casp8−/−Rip3−/−小鼠的拯救(Kaiser et al., 2011;Oberst et al., 2011;Zhang et al., 2011)清楚地表明,在没有Rip3D161N/D161N突变并发症的casp8缺陷胚胎妊娠中期死亡中,早衰RIP3酶活性的贡献(Newton et al., 2014)。 本研究描述了 Rip3K51A/K51A 激酶失活敲入小鼠的构建和表征,这些小鼠存活且可育,旨在证明 RIP3 激酶活性对生命并非必需,并为无毒激酶抑制剂的效果提供模型。[1] |
| 酶活实验 |
RIP3高通量筛选荧光极化(FP)试验用于筛选与RIP3激酶结构域荧光标记探针(GSK ' 657)结合竞争的小分子化合物文库(Pope et al., 1999)。文库化合物抑制RIP3激酶活性的能力是通过使用ADP-Glo测量ATP消耗的实验来评估的(Li et al., 2009)。编码库技术筛选按照前面描述进行(Deng et al., 2012)。kinome面板的体外分析由Reaction Biology Corporation使用“HotSpot”分析平台进行(Anastassiadis等,2011)。Kinome树表示是使用Kinome Mapper生成的。[1]
通过结合和激酶抑制实验,以纯化的、杆状病毒表达的重组人 RIP3 激酶结构域(氨基酸 2-328)为靶标,筛选小分子抑制剂候选物 [1]。 使用荧光偏振法测定化合物与 RIP3 激酶结构域的结合亲和力 [1]。 使用 ADP-Glo 法测定化合物对重组 RIP3 激酶活性的抑制效力 [1]。 通过在 1 μM 浓度下测试化合物对 300 种人类蛋白激酶组的选择性进行 profiling [1]。 |
| 细胞实验 |
细胞活力、caspase活性和显微观察[1]
使用Cell Titer-Glo发光细胞活力测定试剂盒间接检测ATP,使用细胞毒性LDH测定试剂盒检测乳酸脱氢酶释放,使用IncuCyte对SYTOX Green的摄取。利用caspase - glo 3/7活性测定系统和caspase - glo 8活性测定系统分别测定效应物caspase活性。如前所述(Tandon和Mocarski, 2008)进行透射电子显微镜(TEM),并在Emory electron microscopy Core使用JEOL JEM-1400透射电子显微镜获得图像。 细胞活力测定[1] 将L929细胞(5000细胞/孔)、BMDM细胞(30000细胞/孔)、NIH3T3细胞(10000细胞/孔)、3T3-SA细胞(10000细胞/孔)、SVEC4-10细胞(10000细胞/孔)分别接种到康宁96孔组织培养板(3610)中。在大多数实验中,根据制造商的说明,使用cell Titer-Glo发光细胞活力测定试剂盒通过测量细胞内ATP水平来评估细胞活力,并将结果与对照培养物相比较。 坏死性凋亡抑制实验:用刺激剂处理人 HT-29 细胞或小鼠细胞,并在不同浓度的 GSK'843 存在下培养。处理 18-24 小时后,通常通过测量细胞 ATP 水平来评估细胞活力 [1]。 凋亡诱导实验:用高浓度 GSK'843 处理各种细胞系,并在存在或不存在 caspase 抑制剂的情况下培养。通过活力测定、显微镜检查膜通透性、流式细胞术检测 caspase-3 切割或测量 caspase-3/7 和 caspase-8 酶活性来监测细胞死亡 [1]。 复合物形成的生化分析:在用 GSK'843 处理后,对细胞裂解物中的 FADD 或 FLAG 标记的 RIP3 进行免疫沉淀,然后对 RIP3、RIP1、FADD、Casp8 和 cFLIP 等组分进行免疫印迹分析 [1]。 基因需求验证:使用基因敲除细胞、激酶失活突变细胞、siRNA/shRNA 介导的敲低以及重建实验来验证特定基因在药物作用中的必要性 [1]。 病毒抑制剂作用测试:在用 GSK'843 处理前,用表达 MCMV vIRA 或其突变体的逆转录病毒转导细胞 [1]。 |
| 动物实验 |
小鼠、感染和器官获取 [1]
RIP3K51A/K51A 小鼠和 RIP1K45A/K45A 小鼠(Berger 等,2014)由 Genoway 公司(法国里昂)构建。Rip3-/-(Newton 等,2004)、Tnf-/-(Pasparakis 等,1997)、Rip3-/- Casp8-/-(Kaiser 等,2011)、Rip1-/- Rip3-/- Casp8-/- 和 Rip1-/- Rip3+/- Casp8-/-(Kaiser 等,2014)小鼠已有相关描述。C57BL/6 小鼠购自 Jackson Laboratory,Rip3-/- 小鼠(Ripk3tm1Vmd)购自 Genentech 公司(Newton 等,2004)。野生型 MCMV K181 株以及 M45mutRHIM 株和表达 lacZ 的 RM461 株此前已有描述(Stoddart 等,1994;Upton 等,2010)。小鼠腹腔注射 10⁶ PFU MCMV M45mutRHIM 株。感染后 14 天,小鼠再次腹腔注射表达 lacZ 的 MCMV 株 RM427,4 天后采集器官。器官病毒滴度测定方法如前所述(Upton 等,2010)。 Rip3K51A/K51A 激酶失活敲入小鼠的构建 [1] 敲入策略由 genOway 公司设计并实施。Rip3 基因靶向载体由 C57BL/6 小鼠基因组 DNA 构建。 K51A 点突变插入 Rip3 外显子 2,而新霉素抗性基因盒插入内含子 3(两侧为 FRT 位点,以便进一步通过 Flp 介导的切除)。包含 K51A 点突变的外显子 2 两侧为 loxP 位点,可通过 Cre 介导的重组实现组成型或条件性缺失。 本手稿未描述涉及化合物 GSK'843 给药的具体动物实验。体内结论基于对基因工程改造的 Rip3K51A/K51A 敲入小鼠的表型研究 [1]。 Rip3K51A/K51A 小鼠的构建采用胚胎干细胞的标准基因靶向技术,然后通过繁殖获得纯合突变体。本研究使用小鼠进行活力/生育力评估、细胞分离(MEF、BMDM)以及MCMV感染模型构建[1]。 对于MCMV感染模型,将不同基因型(WT、Rip3/-、Rip3K51A/K51A)的同龄小鼠腹腔注射MCMV-M45mutRHIM病毒。感染后3天,通过噬斑试验测定脾脏和肝脏中的病毒滴度[1]。 为了进行免疫应答分析,先用MCMV-M45mutRHIM病毒对小鼠进行免疫,随后用表达lacZ的MCMV病毒进行攻击。攻击后4天收集脾细胞,并用M45特异性肽刺激,通过细胞内细胞因子染色检测CD8+ T细胞中IFNγ和TNFα的产生[1]。 |
| 毒性/毒理 (Toxicokinetics/TK) |
本研究中观察到的GSK'843的主要毒性是其在高浓度(≥3 μM)下可浓度依赖性地诱导细胞培养物中caspase-8依赖性细胞凋亡,这是一种与RIP3构象变化和RHIM信号通路激活相关的靶向效应[1]。
|
| 参考文献 | |
| 其他信息 |
受体相互作用蛋白激酶3 (RIP3 或 RIPK3) 已成为坏死性凋亡的核心参与者,也是控制炎症性疾病的潜在靶点。本文研究表明,三种选择性小分子化合物能够抑制 RIP3 激酶依赖性坏死性凋亡,但令人惊讶的是,它们会浓度依赖性地诱导细胞凋亡,这削弱了它们的治疗价值。这些化合物与 RIP3 相互作用,通过 RHIM 驱动的 RIP1 (RIPK1) 募集激活 caspase 8 (Casp8),从而组装成 Casp8-FADD-cFLIP 复合物,该过程完全独立于促坏死激酶活性和 MLKL。RIP3 激酶失活的 D161N 突变体能够诱导自发性细胞凋亡,而无需化合物参与;而 D161G、D143N 和 K51A 突变体与野生型一样,只有在化合物存在的情况下才会触发细胞凋亡。因此,RIP3-K51A突变小鼠(Rip3(K51A/K51A))具有生存能力和生育能力,这与Rip3(D161N/D161N)小鼠的围产期致死形成鲜明对比。RIP3通过类似Ripoptosome的平台维持坏死性凋亡和细胞凋亡的平衡。这项工作揭示了一种共同机制,即通过治疗或基因手段扰乱RIP3来揭示RHIM驱动的细胞凋亡。[1]
Toll样受体(TLR)信号通路由病原体相关分子模式触发,这些模式介导已知的细胞因子驱动通路,激活NF-κB以及IRF3/IRF7。此外,TLR3驱动caspase 8调控的程序性细胞死亡通路,类似于TNF家族死亡受体信号通路。我们发现,在刺激 TLR2、TLR3、TLR4、TLR5 或 TLR9 的过程中抑制或消除 caspase 8 会导致受体相互作用蛋白 (RIP) 3 激酶依赖性的程序性坏死,该过程通过含 TIR 结构域的衔接蛋白诱导干扰素-β (TRIF) 或 MyD88 信号转导途径发生。TLR3 或 TLR4 通过 TRIF 与 RIP3 激酶(也称为 RIPK3)的 RIP 同型相互作用基序依赖性结合直接激活程序性坏死。在成纤维细胞中,该通路不依赖于 RIP1 或其激酶活性,但仍依赖于 RIP3 激酶下游的混合谱系激酶结构域样蛋白 (MLKL)。本文描述了两种小分子RIP3激酶抑制剂,并利用它们证明了RIP3激酶在RIP1-RIP3、DAI-RIP3和TRIF-RIP3复合物诱导的程序性坏死中的共同作用。TLR信号通路后的细胞命运决定与死亡受体信号通路平行,并依赖于caspase 8来抑制RIP3依赖性程序性坏死,无论该坏死是由TRIF-RIP3-MLKL通路直接启动,还是通过TNF激活和RIP1-RIP3-MLKL坏死性凋亡通路间接启动。[2] GSK'843是一种选择性小分子RIP3激酶抑制剂,与GSK'872一起从常规小分子化合物库筛选中被发现[1]。 该研究揭示了RIP3的双重作用。 RIP3蛋白的激酶活性驱动MLKL依赖性坏死性凋亡,而RIP3蛋白本身也能作为促凋亡复合物的支架。高浓度的GSK'843(以及其他RIP3抑制剂)可诱导RIP3发生构象变化,促进RHIM依赖性寡聚化,并募集RIP1、FADD、cFLIPL和Casp8,最终导致不依赖于激酶活性的细胞凋亡[1]。这种促凋亡活性对开发RIP3激酶抑制剂作为抗炎疗法构成潜在挑战,因为它可能代表一种不良的靶向毒性[1]。本研究区分了各种RIP3激酶失活突变体。D161N突变可自发诱导细胞凋亡(模拟高剂量抑制剂的作用),而K51A突变则不会,除非存在抑制剂。这表明激酶活性丧失本身并不具有毒性,但特定的扰动(某些突变或高浓度抑制剂结合)可以将RIP3转化为促凋亡接头蛋白[1]。 Rip3K51A/K51A小鼠的存活率证明了体内消除RIP3激酶活性本身并不致命,也不会损害免疫功能,这表明如果RIP3抑制剂能够避免触发促凋亡的构象变化,那么无毒的RIP3抑制剂是可行的[1]。 |
| 分子式 |
C₁₉H₁₅N₅S₂
|
|---|---|
| 分子量 |
377.49
|
| 精确质量 |
377.076
|
| 元素分析 |
C, 60.46; H, 4.01; N, 18.55; S, 16.99
|
| CAS号 |
1601496-05-2
|
| 相关CAS号 |
1601496-05-2
|
| PubChem CID |
91885439
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.5±0.1 g/cm3
|
| 沸点 |
640.0±55.0 °C at 760 mmHg
|
| 闪点 |
340.8±31.5 °C
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
| 折射率 |
1.832
|
| LogP |
4.9
|
| tPSA |
126
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
522
|
| 定义原子立体中心数目 |
0
|
| SMILES |
S1C=C(C2C=CC3=C(C=2)N=CS3)C2C(N)=NC=C(C3=CC(C)=NN3C)C1=2
|
| InChi Key |
BPKSNNJTKPIZKR-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C19H15N5S2/c1-10-5-15(24(2)23-10)12-7-21-19(20)17-13(8-25-18(12)17)11-3-4-16-14(6-11)22-9-26-16/h3-9H,1-2H3,(H2,20,21)
|
| 化学名 |
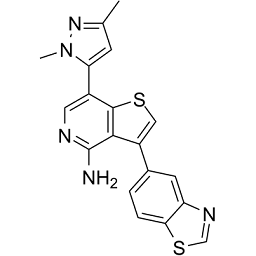
3-(1,3-benzothiazol-5-yl)-7-(2,5-dimethylpyrazol-3-yl)thieno[3,2-c]pyridin-4-amine
|
| 别名 |
GSK843; GSK-843; GSK 843; 1601496-05-2; GSK-843; GSK843; GSK'843; 3-(1,3-benzothiazol-5-yl)-7-(1,3-dimethyl-1H-pyrazol-5-yl)thieno[3,2-c]pyridin-4-amine; CHEMBL4441118; 3-(1,3-BENZOTHIAZOL-5-YL)-7-(2,5-DIMETHYLPYRAZOL-3-YL)THIENO[3,2-C]PYRIDIN-4-AMINE; 3-(benzo[d]thiazol-5-yl)-7-(1,3-dimethyl-1H-pyrazol-5-yl)thieno[3,2-c] pyridin-4-amine; . GSK'843
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~50 mg/mL (~132.5 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 4.8 mg/mL (12.72 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 48.0mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (5.51 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6491 mL | 13.2454 mL | 26.4908 mL | |
| 5 mM | 0.5298 mL | 2.6491 mL | 5.2982 mL | |
| 10 mM | 0.2649 mL | 1.3245 mL | 2.6491 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
