| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
KD: 7.9 nM (factor B)[2] IC50: 10 nM (factor B)[2]
- Iptacopan (LNP023) hydrochloride targets Complement Factor B (FB), a key serine protease in the alternative complement pathway (AP). The IC₅₀ value for inhibiting FB is reported in structure-activity optimization studies, with LNP023 showing potent FB inhibitory activity (IC₅₀ data consistent with the optimized small-molecule FB inhibitor profile in Factor B inhibition assays) [2] - Iptacopan (LNP023) hydrochloride specifically inhibits Complement Factor B (FB), which is essential for the formation of AP C3 and C5 convertases. High-throughput screening (HTS) and X-ray cocrystal structure-guided optimization confirmed its selective binding to FB, with inhibitory potency optimized to meet preclinical therapeutic requirements [3] |
|---|---|
| 体外研究 (In Vitro) |
1. 抑制C3肾小球病(C3G)患者血清中的C3转化酶活性:将LNP023 以1.1 µM和3.3 µM的浓度与C3G患者血清共孵育,可有效阻断AP介导的C3降解(通过免疫固定电泳检测)。该抑制效果与阳性对照(EDTA,可完全阻断AP)相当,且优于仅阻断经典/凝集素途径的对照(Mg²⁺-EGTA,不影响AP) [2]
2. 防止人阵发性睡眠性血红蛋白尿症(PNH)红细胞溶血:将LNP023 以1 µM的浓度与PNH患者红细胞(来自3例PNH患者,重复检测2~14次)及患者血清共孵育,可显著降低红细胞溶血率。剂量效应曲线显示,LNP023 对PNH红细胞溶血的抑制活性与因子D(FD)抑制剂、抗C5抗体相当 [2] 3. 抑制肾炎因子(nephritic factor)稳定的C3转化酶活性:将包被有预形成C3转化酶的绵羊红细胞与C3G患者血清IgG(用于稳定C3转化酶)及0.15 µM的LNP023 共孵育。结果显示,LNP023 可显著降低肾炎因子稳定的C3转化酶的稳定性(以% Z = Z₁₅min/Z₀min × 100计算),证实其可干扰AP C3转化酶的功能 [2] 4. 选择性抑制FB:LNP023 经HTS筛选发现,结合FB-抑制剂复合物的X射线共晶结构进行优化,对FB的选择性显著高于其他丝氨酸蛋白酶,对经典/凝集素途径的蛋白酶无明显脱靶抑制,明确其AP特异性抑制特征 [3] 在 50% 的人血清中,iptacopan (LNP023) 可有效防止补体旁路途径 (AP) 引起的膜攻击复合物 (MAC) 的形成(IC50 值:130 nM)[2]。在 41 种人类蛋白酶中,iptacopan (LNP023) 的 IC50 值 >30 μM,与其他蛋白酶(包括 AP 蛋白因子 D (>100 μM))相比具有优异的选择性 [3]。 |
| 体内研究 (In Vivo) |
1. 在KRN诱导的关节炎小鼠模型(C57BL/6小鼠)中的疗效:首次给药1 h后,通过注射KRN/I-Ag7血清诱导关节炎,LNP023 以20 mg/kg、60 mg/kg、180 mg/kg的剂量每日两次(b.i.d.)口服给药(每组n=8),结果显示:
- 与溶剂对照组相比,LNP023 可剂量依赖性降低疾病评分(通过关节肿胀评估);
- 关节组织中AP激活产物(Ba、C3d、C5a)的水平显著降低;
- 组织学改善:给药6天后,炎症细胞浸润减少、骨侵蚀减轻、蛋白多糖保留增加(通过H&E和番红O染色评估),差异具有统计学意义(与溶剂组相比,P < 0.001,P < 0.0001) [2]
2. 在被动Heymann肾炎大鼠模型(膜性肾病模型)中的疗效:通过注射抗Fx1A抗体血清诱导膜性肾病,对照组注射正常血清(n=5),LNP023 以20 mg/kg、60 mg/kg的剂量每日两次口服给药,分为两种方案: - 预防性给药(从第0天开始):持续15天,可降低尿蛋白/肌酐比值(UTP/UCREA),并防止肾小球病变(肾小球增大、基底膜增厚)和肾小管变性(通过H&E染色评估); - 治疗性给药(从疾病诱导后第6天开始):持续8天,可显著降低已形成的蛋白尿,并减少肾小球C3沉积(组织学评分,代表性染色显示C3沉积较溶剂组减少),差异具有统计学意义(与溶剂组相比,P < 0.05,P < 0.01,P < 0.001;每组n=9) [2] 在大鼠膜性肾病实验模型中,diptacopan(LNP023;20-180 mg/kg;口服)在预防和治疗剂量下均有效,并可预防 KRN (150 μL) 诱导的小鼠关节炎 [2]。口服给药后(狗 10 mg/kg,大鼠30毫克/千克)[3]。 ?Iptacopan 是由静脉内给药(狗 1.0 mg/kg,大鼠 0.1 mg/kg)后的大分布体积(2.3 和 0.6 L/kg)和高血浆清除率(分别为 8 和 2 mL/min/kg)引起的[3]。 |
| 酶活实验 |
1. 补体因子B(FB)丝氨酸蛋白酶活性实验:将重组人FB与特异性肽底物(对应AP级联中FB的切割位点)在FB活性优化缓冲液中孵育,加入系列浓度(亚纳摩尔至微摩尔级)的LNP023,37°C孵育特定时间后,通过检测特定波长下的吸光度量化切割底物的生成量(反映FB酶活性)。通过FB活性抑制率与LNP023 浓度的剂量效应曲线计算IC₅₀,证实LNP023 对FB具有强效、可逆的抑制活性 [2]
2. FB结合实验(X射线晶体学指导):将纯化人FB与LNP023 形成复合物并结晶,收集X射线衍射数据(初始FB-抑制剂复合物的分辨率可达1.64 Å)以解析结合模式。结果显示,LNP023 结合于FB的活性中心,与催化残基(His57、Ser195)及S1口袋关键残基相互作用,这是其抑制特异性的核心机制,该结合模式为LNP023 potency和选择性的进一步优化提供了依据 [2, 3] 体外抑制试验[2] 通过使用CVF:Bb作为C3转化酶的稳定替代物和纯化的内源性C3作为底物,或者通过使用FB和Cy5标记的小分子抑制剂作为探针的竞争结合测定来测试化合物的FB抑制。通过酵母多糖A诱导的MAC形成,在50%的人血清或50%的人全血中测量AP抑制。将血清或全血与化合物预孵育30分钟,然后转移到酵母多糖A包被的平板上。通过ELISA用抗C9新表位抗体检测MAC形成。以类似的方式测量小鼠血清中的AP补体沉积,不同之处在于检测C3b沉积而不是MAC形成。SI附录中提供了有关蛋白质纯化和所有体外测定的更多详细信息。 |
| 细胞实验 |
1. PNH红细胞溶血实验:从PNH患者血液中分离红细胞并洗涤去除血浆成分,将红细胞重悬于含Mg²⁺(支持AP激活)的缓冲液中,加入PNH患者血清(提供补体成分)及系列浓度(0.1~10 µM)的LNP023,37°C孵育固定时间后,通过流式细胞术(FACS)检测红细胞膜完整性标志物,量化溶血率。结果证实LNP023 可剂量依赖性抑制PNH红细胞溶血,1 µM浓度时即具有显著疗效 [2]
2. 绵羊红细胞C3转化酶实验:将绵羊红细胞与纯化C3b、FB在FD存在下孵育,包被形成AP C3转化酶(C3bBb);随后加入0.15 µM的LNP023 及C3G患者血清IgG(稳定C3转化酶),37°C孵育15 min后,通过检测C3切割产物量化活性C3转化酶的量。结果显示LNP023 可降低肾炎因子稳定的C3转化酶的稳定性,证实其在细胞体系中可靶向AP C3转化酶功能 [2] |
| 动物实验 |
动物/疾病模型: KRN诱导关节炎的C57BL/6小鼠[2]
剂量: 20、60和180 mg/kg:灌胃给药(po);每日两次(bid),持续14天 实验结果: 阻断KRN诱导的关节炎。 1. KRN诱导的关节炎小鼠模型(C57BL/6小鼠): - 疾病诱导:小鼠在首次给予LNP023 1小时后注射KRN/I-Ag7血清以诱导关节炎; - 给药方案:LNP023以20 mg/kg、60 mg/kg和180 mg/kg的剂量每日两次(bid)灌胃给药,持续6天。设置溶剂对照组(每组n=8只小鼠); - 样本采集:第 6 天处死小鼠。采集关节组织用于补体激活产物(Ba、C3d、C5a)的测定和组织学分析(H&E 和番红 O 染色)[2] 2. 被动性 Heymann 肾炎大鼠模型(Sprague-Dawley 大鼠): - 疾病诱导:给大鼠注射抗 1A 组分(抗 Fx1A)抗体血清以诱导膜性肾病。对照组注射正常血清(n=5); - 给药方案: - 预防性给药:从第 0 天开始,每天两次口服 LNP023,剂量分别为 20 mg/kg 和 60 mg/kg(每组 n=9),持续 15 天; - 治疗剂量:从第6天(疾病发作后)开始,以20 mg/kg和60 mg/kg的剂量,每日两次口服LNP023,持续8天(每组n=9);- 样本采集:定期收集尿液,测定尿蛋白/肌酐比值(UTP/UCREA)。在第14天(治疗组)或第15天(预防组),处死大鼠,并收集肾脏组织进行组织学分析(肾小球病变、肾小管变性)和肾小球C3沉积评估[2] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服后,伊普他考泮的血浆浓度峰值约为给药后2小时。按照推荐的每日两次、每次200毫克的给药方案,大约5天即可达到稳态,且药物蓄积量较小(1.4倍)。一项针对健康志愿者的食物影响研究表明,高脂饮食对伊普他考泮的暴露量没有产生具有临床意义的影响。 在一项人体研究中,单次口服100毫克[14C]-伊普他考泮后,放射性物质(伊普他考泮及其代谢物)的平均总排泄量为:粪便中71.5%,尿液中24.8%,总平均排泄量超过给药剂量的96%。具体而言,17.9% 的剂量以原药伊普他考潘的形式经尿液排出,16.8% 的剂量以原药伊普他考潘的形式经粪便排出。 每日两次服用 200 mg 伊普他考潘后,稳态表观分布容积约为 288 L。 每日两次服用 200 mg 伊普他考潘后,稳态清除率为 7.96 L/h。 代谢/代谢物 代谢是伊普他考潘的主要消除途径,约 50% 的剂量归因于氧化途径。伊普他考潘的代谢包括 N-去烷基化、O-去乙基化、氧化和脱氢,主要由 CYP2C8 驱动 (98%),CYP2D6 的贡献较小 (2%)。伊普他考泮主要通过UGT1A1、UGT1A3和UGT1A8的葡萄糖醛酸化作用进行II期代谢。在血浆中,伊普他考泮是主要成分,占药物相关物质的83%。血浆中仅检测到两种酰基葡萄糖醛酸苷代谢物,含量较低,分别占药物相关物质的8%和5%。伊普他考泮代谢物不具有药理活性。 生物半衰期 每日两次服用200 mg伊普他考泮后,其稳态半衰期(t1/2)约为25小时。 |
| 毒性/毒理 (Toxicokinetics/TK) |
妊娠期和哺乳期用药
◉ 哺乳期用药概述 目前尚无关于哺乳期使用伊普他可泮的信息。生产商指出,在治疗期间以及末次给药后5天内应停止哺乳。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对哺乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白结合 伊普他可泮的血浆蛋白结合率呈浓度依赖性,这是由于其与体循环中的靶因子B结合所致。在相关的临床血浆浓度下,体外伊普他可泮的蛋白结合率为75%至93%。 |
| 参考文献 |
|
| 其他信息 |
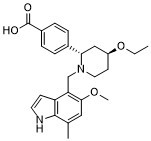
伊普他可泮(Iptacopan)属于吲哚类化合物,其结构为1H-吲哚,在4、5和6位分别被[(2S,4S)-2-(4-羧基苯基)-4-乙氧基哌啶-1-基]甲基、甲氧基和甲基取代。它是一种强效的补体因子B抑制剂(IC50 = 10 nM),并具有潜在的免疫调节活性。它既是补体因子B抑制剂,也是免疫调节剂。伊普他可泮属于苯甲酸类、哌啶类、醚类、吲哚类、二醚类、单羧酸类和叔胺类化合物。它是iptacopan(1+)的共轭碱。
Iptacopan是一种小分子B因子抑制剂,此前曾被研究作为治疗罕见血液疾病阵发性睡眠性血红蛋白尿症(PNH)的潜在药物,其作用机制是通过抑制补体因子B。B因子是替代补体途径的正向调节因子,它能激活C3转化酶,进而激活C5转化酶。这对于PNH尤为重要,因为PNH的疾病标志之一是PIGA基因突变。由于这种突变,所有子代红细胞都将缺乏糖基磷脂酰肌醇锚定蛋白,而这些蛋白通常锚定两种膜蛋白CD55和CD59,从而保护血细胞免受替代补体途径的攻击。此外,iptacopan的优势在于它靶向B因子,仅影响替代补体途径,而不会影响经典补体途径和凝集素途径,从而使机体仍能对病原体产生足够的免疫反应。 2023年12月6日,商品名为Fabhalta的伊普他可潘(Iptacopan)获得美国食品药品监督管理局(FDA)批准,用于治疗成人阵发性睡眠性血红蛋白尿症(PNH)。该批准基于III期APPL-PNH和APPOINT-PNH研究的良好结果,这两项研究分别显示82.3%和77.5%的患者在无需输血的情况下实现了持续的血红蛋白水平改善。 伊普他可潘是一种口服的小分子补体因子B(FB)抑制剂,具有潜在的免疫调节活性。给药后,伊普他可潘与FB结合,阻止替代途径(AP)C3转化酶(C3bBb)的形成。这限制了C3裂解为活性片段C3b,并可能预防某些补体驱动疾病(例如阵发性睡眠性血红蛋白尿症 (PNH))中C3b介导的血管外溶血。 药物适应症 伊普他考泮适用于治疗成人阵发性睡眠性血红蛋白尿症。 阵发性睡眠性血红蛋白尿症的治疗 作用机制 伊普他考泮与替代补体途径的B因子结合,调节C3的裂解、下游效应分子的生成以及末端途径的放大。在阵发性睡眠性血红蛋白尿症中,血管内溶血 (IVH) 由下游膜攻击复合物 (MAC) 介导,而血管外溶血 (EVH) 则由C3b调理作用促进。伊普他可泮通过作用于补体级联反应的替代途径,控制C3b介导的脑室外出血(EVH)和末端补体介导的脑室内出血(IVH)。 药效学 在健康志愿者中,单次服用伊普他可泮后约2小时,替代补体途径生物标志物、体外替代途径检测以及血浆Bb(B因子片段)水平开始受到抑制。在接受抗C5治疗和伊普他可泮(200 mg,每日两次)联合治疗的阵发性睡眠性血红蛋白尿(PNH)患者中,第8天首次观察时,体外替代途径检测和血浆Bb水平较基线分别下降了54.1%和56.1%。在未接受治疗的PNH患者中,接受伊普他可泮(200 mg,每日两次)治疗4周后,首次观察时,这些相同的生物标志物较基线分别下降了78.4%和58.9%。在接受抗C5治疗和每日两次FABHALTA 200 mg治疗的阵发性睡眠性血红蛋白尿症(PNH)患者中,基线时平均PNH红细胞克隆大小为54.8%,13周后增至89.2%;基线时PNH II型和III型红细胞伴C3沉积的比例为12.4%,13周后降至0.2%。在未接受治疗的PNH患者中,基线时平均PNH红细胞克隆大小为49.1%,12周后增至91.1%;由于脑室内出血(IVH)占主导地位,该人群中PNH II型和III型红细胞伴C3沉积的比例可忽略不计。伊普他可泮可降低血清乳酸脱氢酶(LDH)水平。在既往接受过依库珠单抗治疗的阵发性睡眠性血红蛋白尿症 (PNH) 患者中,所有接受每日两次 200 mg FABHALTA 治疗的患者在 13 周时乳酸脱氢酶 (LDH) 水平降至正常值上限 (ULN) 的 1.5 倍以下。在初治 PNH 患者中,每日两次 200 mg 伊普他考泮在 12 周后使 LDH 水平较基线降低 60% 以上,并在 2 年的研究结束时仍维持该疗效。在一项针对健康志愿者的 QTc 间期临床研究中,单次服用高达 1200 mg 的超治疗剂量伊普他考泮(其峰值浓度超过正常值上限的 4 倍)未对心脏复极化或 QT 间期产生影响。<1 小时>1. 作用机制:盐酸伊普他考泮 (LNP023) 抑制补体因子 B (FB),从而阻断替代补体途径 (AP)。通过靶向FB,LNP023可阻止AP C3转化酶(C3bBb)和C5转化酶的形成,从而抑制AP介导的补体激活(例如,C3沉积、MAC组装)和下游病理过程(例如,溶血、炎症、组织损伤)[2, 3] 2. 治疗适应症:LNP023正在开发用于治疗补体介导的疾病,包括阵发性睡眠性血红蛋白尿症(PNH)、C3肾小球病(C3G)、非典型溶血性尿毒综合征(aHUS)、膜性肾病、类风湿性关节炎(以KRN诱导的关节炎为模型)和年龄相关性黄斑变性(AMD)[2, 3] 3. 药物特性:LNP023是一种口服生物利用度高的选择性小分子FB抑制剂。该化合物通过高通量筛选 (HTS) 鉴定,并利用 FB-抑制剂复合物的 X 射线共晶结构信息进行优化,确保了其对 FB 的高效特异性抑制作用 [3] 4. 临床前疗效亮点:在临床前模型中,LNP023 显示出预防和治疗功效(例如,降低肾病大鼠的蛋白尿,减轻关节炎小鼠的关节炎症),并能有效抑制人血清中的 AP 活化(例如,C3G、PNH),支持其临床开发 [2] 5. 文献 [1] (Semin Hematol. 2018) 主要关注 PNH 的补体疗法,但未提及盐酸伊普他考泮 (LNP023) 或其相关特性 [1] |
| 分子式 |
C25H30N2O4
|
|---|---|
| 分子量 |
422.516706943512
|
| 精确质量 |
422.22
|
| 元素分析 |
C, 71.07; H, 7.16; N, 6.63; O, 15.15
|
| CAS号 |
1644670-37-0
|
| 相关CAS号 |
Iptacopan hydrochloride;1646321-63-2
|
| PubChem CID |
90467622
|
| 外观&性状 |
Off-white to gray solid powder
|
| LogP |
1.8
|
| tPSA |
74.8Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
594
|
| 定义原子立体中心数目 |
2
|
| SMILES |
CCO[C@H]1CCN([C@@H](C1)C2=CC=C(C=C2)C(=O)O)CC3=C(C=C(C4=C3C=CN4)C)OC
|
| InChi Key |
RENRQMCACQEWFC-UGKGYDQZSA-N
|
| InChi Code |
InChI=1S/C25H30N2O4/c1-4-31-19-10-12-27(22(14-19)17-5-7-18(8-6-17)25(28)29)15-21-20-9-11-26-24(20)16(2)13-23(21)30-3/h5-9,11,13,19,22,26H,4,10,12,14-15H2,1-3H3,(H,28,29)/t19-,22-/m0/s1
|
| 化学名 |
4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid
|
| 别名 |
LNP023; Iptacopan; LNP-023; LNP 023; Fabhalta
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~118.34 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 5 mg/mL (11.83 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 50.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 5 mg/mL (11.83 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 50.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 5 mg/mL (11.83 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3668 mL | 11.8338 mL | 23.6675 mL | |
| 5 mM | 0.4734 mL | 2.3668 mL | 4.7335 mL | |
| 10 mM | 0.2367 mL | 1.1834 mL | 2.3668 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy
CTID: NCT04820530
Phase: Phase 3 Status: Completed
Date: 2024-10-09
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
