| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|
| 体外研究 (In Vitro) |
甲苯咪唑(1 nM-0.1 mM;72 小时)抑制 GL261 肿瘤神经细胞,IC50 值为 0.24 μM [1]。 0.1 μM 和 1 μM 甲苯咪唑作用 24 小时会导致 060919 多形性星形胶质细胞破坏。甲苯咪唑(10 nM–10 μM;48 小时)抑制肿瘤 (GBM) 细胞中的微管聚合和微管结构,抑制 Hh 信号传导,并减少下游 Hh 蛋白表达。它还通过降低肿瘤组织中的 Gli1 来调节 Hh 蛋白的表达 [1]。 Gli1 表达受甲硝唑抑制,IC50 值为 516 nM[2]。当暴露于甲苯咪唑(10 nM-10 μM;48 小时)时,具有组成型 Hh 激活的人髓母细胞瘤肿瘤细胞表现出表达降低并阻碍初级纤毛发育。当甲苯咪唑和维莫德吉联合使用时,可以实现对正常 Hh 信号传导的加性抑制 [2]。在治疗环境中,甲苯咪唑已被证明可以成功治疗严重的中枢神经系统包虫病病例。荧光成像 [1]
|
|---|---|
| 体内研究 (In Vivo) |
对人多形性母细胞瘤 (GBM) 异种移植物 060919 和同基因 GL261 小鼠模型均给予甲苯咪唑(50 mg/kg;口服;前 20 天每天一次,每周 5 天,休息两天;45 天)。
|
| 细胞实验 |
免疫荧光 [1]
细胞类型: 多形性胶质母细胞瘤 (GBM) 060919 细胞 测试浓度: 1 μM 孵育时间:24小时 实验结果:微管结构被破坏。系统渗透特性[3]。 免疫荧光 [2] 细胞类型: DAOY 和 hTERT-RPE1 细胞 测试浓度: 0、0.1、0.5、0.75 和1 μM 孵育持续时间:12 小时 实验结果:GLI1 蛋白水平降低,caspase-3 蛋白水平增加裂解。 |
| 动物实验 |
动物/疾病模型: C57BL/6 小鼠(5-6 周龄),植入 GL261 胶质瘤细胞和 060919 人胶质母细胞瘤 (GBM) [1]
剂量: 50 mg/kg;抑制小鼠模型颅内肿瘤生长 [1]。使用 50% (v/v) 芝麻油和 PBS [2] 给药途径: 灌胃;肿瘤植入后 5 天开始,前 20 天每日剂量为 50 mg/kg,之后改为 50 mg/kg,持续 5 天,每周停药 2 天。 实验结果: 在同源 GL261 小鼠模型中,平均生存期从对照组的 30 天延长至 49 天。在 060919 人类胶质母细胞瘤 (GBM) 异种移植小鼠模型中,平均生存期延长至 65 天,而对照组为 48 天。 |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
本品在胃肠道吸收不良(约5%~10%)。脂肪类食物可增加吸收。 在人体内,约2%的甲苯咪唑经尿液排出,其余部分以原药或其主要代谢物的形式经粪便排出。 排泄:粪便:约95%以原药或主要代谢物(2-氨基衍生物)的形式经粪便排出。肾脏:约2%~5%以原药或主要代谢物的形式经尿液排出。 血清峰浓度:每日两次,每次100 mg,连续服用3天后:甲苯咪唑:不超过0.03 μg/mL。2-氨基代谢物:不超过0.09 μg/mL。据报道,长期高剂量治疗中血清浓度可达 0.5 μg/mL。 血清峰浓度出现时间:2 至 5 小时(范围:0.5 至 7 小时)。 甲苯咪唑与血浆蛋白高度结合。目前尚不清楚甲苯咪唑是否会分泌到乳汁中。 有关甲苯咪唑(共 11 项)的更多吸收、分布和排泄(完整)数据,请访问 HSDB 记录页面。 代谢/代谢物 主要在肝脏代谢。主要代谢物为 2-氨基-5-苯甲酰基苯并咪唑,但也会代谢为无活性的羟基和羟基氨基代谢物。所有代谢物均无驱虫活性。 主要在肝脏代谢;代谢为无活性的氨基、羟基和羟基氨基代谢物;主要代谢产物是2-氨基-5-苯甲酰基苯并咪唑。 尽管甲苯咪唑的确切代谢途径尚未完全确定,但该药物通过脱羧代谢为2-氨基-5(6)-苯并咪唑基苯基酮;该代谢产物不具有驱虫活性。 甲苯咪唑……代谢广泛。两种主要代谢产物,即5-(α-5-羟基苄基)-2-苯并咪唑氨基甲酸酯和2-氨基-5-苯甲酰基苯并咪唑,其清除率低于甲苯咪唑本身。甲苯咪唑而非其代谢产物似乎是活性药物形式。甲苯咪唑及其代谢物的结合物已在胆汁中发现,但尿液中几乎没有未代谢的甲苯咪唑。 生物半衰期 肝功能正常患者的生物半衰期为2.5至5.5小时(范围2.5至9小时)。肝功能受损(胆汁淤积)患者的生物半衰期约为35小时。 肝功能正常:2.5至5.5小时(范围:2.5至9小时)。肝功能受损(胆汁淤积):约35小时。 据报道,甲苯咪唑的消除半衰期约为2.8-9小时。 |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
甲苯咪唑在常规剂量下通常不会引起血清酶升高,尽管疗程通常较短,且鲜有监测酶升高的报道。高剂量治疗(由于阿苯达唑的出现,目前已很少使用)可导致血清转氨酶水平升高(正常值的2至10倍),但通常耐受性良好。有罕见的甲苯咪唑引起急性肝损伤的报道,尤其是在重复给药或高剂量给药的情况下。起病通常在开始或重新开始治疗后的几天内,表现为发热和不适。血清酶升高的模式通常为肝细胞性,黄疸不常见。停药后,这些异常通常会迅速消退。过敏反应的典型表现(皮疹、发热和嗜酸性粒细胞增多)以及肝活检可能显示肉芽肿。 可能性评分:D(长期治疗可能导致临床上明显的肝损伤)。 妊娠和哺乳期用药 ◉ 哺乳期用药概述 甲苯咪唑很少分泌到乳汁中,口服吸收也很差。关于哺乳期使用甲苯咪唑的报告显示,母乳喂养的婴儿未出现不良反应。有少数病例报告称,使用甲苯咪唑后乳汁分泌减少,但没有确凿的证据表明这是由该药物引起的。无需特别注意。 ◉ 对母乳喂养婴儿的影响 一项病例系列报告了45名哺乳期妇女,她们服用甲苯咪唑,剂量范围为每日一次100毫克至每日两次200毫克,持续3天。约一半的婴儿服用100毫克,7至14天后重复服用一次。33名婴儿为纯母乳喂养,年龄在1至30周之间。12名部分母乳喂养的婴儿中有8名超过20周龄。所有婴儿的母亲均未报告任何不良反应。 在刚果民主共和国,研究人员对33名由住院服用硝呋莫司的母亲哺乳的婴儿(哺乳程度未说明)进行了随访。30名母亲完成了30剂口服硝呋莫司(15毫克/公斤/天)的疗程,所有母亲均接受了14剂静脉注射依氟鸟氨酸(400毫克/公斤/天),疗程7天,用于治疗人类非洲锥虫病(昏睡病)。17名哺乳期母亲还服用了甲苯咪唑。所有母乳喂养的婴儿均未报告严重不良事件。 ◉ 对泌乳和母乳的影响 一位产后13周的哺乳期母亲正在服用甲硝唑,每次250毫克,每日三次。乳汁分泌似乎未受影响。在治疗的第八天,她排出了一条蛔虫。甲硝唑停用,开始服用甲苯咪唑,每次100毫克,每日两次。患者在排出蛔虫后的几天内感到“紧张”。在甲苯咪唑治疗的第二天,乳汁分泌明显下降,她开始添加配方奶。到第七天,乳汁分泌完全停止。作者认为甲苯咪唑可能是导致乳汁分泌下降的原因,但除了时间关系外,没有提供进一步的证据。 四名患者从产后第一天开始,服用甲苯咪唑,每次100毫克,每日两次,疗程3天。其中2只感染了蛲虫(Enterobius),1只感染了蛔虫(Ascaris),1只感染了钩虫(Ancyclostomia)。所有受试者均成功进行母乳喂养。 一位作者报告称,其通过与生产商的私人沟通获得的信息显示,哺乳期母亲单次口服100毫克甲苯咪唑后未观察到泌乳抑制(未说明产后时间)。 在一项病例系列研究中,45名哺乳期母亲服用甲苯咪唑,剂量范围为每日一次100毫克至每日两次200毫克,持续3天。其中一位母亲报告乳汁分泌量略有下降。 蛋白结合率 90-95% 药物相互作用 初步证据表明,西咪替丁会抑制甲苯咪唑的代谢,并可能导致血浆药物浓度升高。 有限的数据表明,卡马西平和苯妥英钠均可能增强甲苯咪唑的代谢,可能是通过诱导肝微粒体酶,从而导致血浆甲苯咪唑浓度降低。这种相互作用在接受治疗的患者中不太可能具有临床意义。甲苯咪唑可用于治疗肠道蠕虫感染;然而,对于接受甲苯咪唑治疗肠外感染(例如包虫病)的患者,若同时服用卡马西平或苯妥英钠,可能会影响其疗效。在获得更多数据之前,对于接受甲苯咪唑治疗肠外感染的患者,应考虑使用其他抗惊厥药物(例如丙戊酸)。 非人类毒性值 绵羊口服LD50大于80 mg/kg |
| 参考文献 |
[1]. Bai RY, et al. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro Oncol. 2011 Sep;13(9):974-82.
[2]. Larsen AR, et al. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol Cancer Ther. 2015 Jan;14(1):3-13. [3]. Erdinçler P, et al. The role of mebendazole in the surgical treatment of central nervous system hydatid disease. Br J Neurosurg. 1997 Apr;11(2):116-20. |
| 其他信息 |
治疗用途
MeSH标题:抗线虫药 甲苯咪唑适用于治疗由鞭虫(Trichuris trichiura)引起的鞭虫病。/已包含在美国产品标签中/ 甲苯咪唑适用于治疗多种肠道线虫感染。/已包含在美国产品标签中/ 甲苯咪唑适用于治疗由蛲虫(Enterobius vermicularis)引起的蛲虫病。 /包含于美国产品标签/ 有关甲苯咪唑(共19种)的更多治疗用途(完整)数据,请访问HSDB记录页面。 药物警告 长期使用甲苯咪唑治疗期间,应定期评估器官系统功能(包括造血和肝脏功能)。 接受甲苯咪唑治疗的患者罕见的其他不良反应包括脱发、皮疹、瘙痒、荨麻疹、血管性水肿、潮红、呃逆、咳嗽、乏力、嗜睡、寒战、低血压、癫痫发作、肝功能检查短暂异常(例如,血清转氨酶、碱性磷酸酶和/或胆红素浓度升高)、肝炎、血尿素氮升高、血红蛋白浓度和/或血细胞比容降低、白细胞减少症、血小板减少症、嗜酸性粒细胞增多症、血尿素氮增多症和血尿素氮增多症。尿柱状症。也有报道称蛔虫可通过口鼻移行。 接受高剂量(例如,每日 30-50 mg/kg)甲苯咪唑治疗肠外感染的患者中,曾报道出现骨髓抑制,表现为中性粒细胞减少症(包括粒细胞缺乏症)和/或血小板减少症;虽然停药后骨髓抑制通常可逆,但死亡病例罕见。 在常用推荐剂量(即每日 100-200 mg)下,甲苯咪唑似乎只会引起极少的副作用。当使用较高剂量(例如,用于治疗肠外感染如包虫病的剂量)时,副作用似乎更常见,并且在某些情况下可能与药物诱导的寄生虫杀灭作用有关。甲苯咪唑治疗期间偶有短暂性腹泻和腹痛,但通常与大量感染和蠕虫排出有关。甲苯咪唑治疗期间偶有恶心、呕吐、头痛、耳鸣、麻木和眩晕的报道。部分患者出现发热,尤其是在接受高剂量治疗肠外感染的患者中。 有关甲苯咪唑(共9条)的更多药物警告(完整)数据,请访问HSDB记录页面。 药效学 甲苯咪唑是一种(合成的)广谱驱虫药。甲苯咪唑的主要作用机制是通过抑制微管蛋白聚合,导致细胞质微管丢失。 |
| 分子式 |
C16H13N3O3
|
|---|---|
| 分子量 |
295.3
|
| 精确质量 |
295.095
|
| CAS号 |
31431-39-7
|
| 相关CAS号 |
Mebendazole-d3;1173021-87-8
|
| PubChem CID |
4030
|
| 外观&性状 |
Off-white amorphous powder
Crystals from acetic acid and methanol |
| 密度 |
1.4±0.1 g/cm3
|
| 熔点 |
288.5°C
|
| 折射率 |
1.703
|
| LogP |
2.83
|
| tPSA |
84.08
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
22
|
| 分子复杂度/Complexity |
423
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O=C(OC)NC1=NC2=CC=C(C(C3=CC=CC=C3)=O)C=C2N1
|
| InChi Key |
OPXLLQIJSORQAM-UHFFFAOYSA-N InChi Code
|
| InChi Code |
InChI=1S/C16H13N3O3/c1-22-16(21)19-15-17-12-8-7-11(9-13(12)18-15)14(20)10-5-3-2-4-6-10/h2-9H,1H3,(H2,17,18,19,21)
|
| 化学名 |
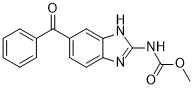
methyl N-(6-benzoyl-1H-benzimidazol-2-yl)carbamate
|
| 别名 |
Mebendazole Vermox Telmin Pantelmin Mebenvet Telmin Vermicol Vermidil Vermin Vermox Wormkuur
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~4.17 mg/mL (~14.12 mM)
H2O : < 0.1 mg/mL |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 0.42 mg/mL (1.42 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 4.2 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 0.42 mg/mL (1.42 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 4.2mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 0.42 mg/mL (1.42 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.3864 mL | 16.9319 mL | 33.8639 mL | |
| 5 mM | 0.6773 mL | 3.3864 mL | 6.7728 mL | |
| 10 mM | 0.3386 mL | 1.6932 mL | 3.3864 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Antileishmanial agent-34
Antileishmanial agent-34
 IID432
IID432
 WEHI-326
WEHI-326
 CYP3A4-IN-5
CYP3A4-IN-5
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
