| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
PD1-PDL1 (IC50 = 6 nM)
Human PD-L1 (Programmed death-ligand 1). [4] |
|---|---|
| 体外研究 (In Vitro) |
在三阴性乳腺癌 (TNBC) 中,PD-1/PD-L1 抑制剂 1 在较低浓度下可使细胞活力降低一半,激活 MDA-MB-231 和 MCF7 中的 ERK,显著增加 MDA-MB-231 和 HCC1806 中的 IL-8 表达,并有效下调 MDA-MB-231 中的 IL-8 表达至一半水平。[2]
BMS-1001 可剂量依赖性地诱导与 PD-L1+ aAPC 共培养的 PD-1 效应细胞中的荧光素酶活性,表明其在细胞界面拮抗 PD-1/PD-L1 检查点,EC50 值在三位数纳摩尔范围内,并且与治疗性抗体相比,最大细胞活化较低。 [4] BMS-1001呈剂量依赖性地消除了可溶性PD-L1 (sPD-L1)对抗CD3介导的Jurkat T细胞活化的抑制作用,在sPD-L1摩尔浓度为2倍和5倍时(终浓度为3 μM,BMS与sPD-L1的摩尔比为5:1),细胞活化完全恢复至单独使用抗CD3的水平。[4] 在流式细胞术中,用BMS-1001预孵育荧光标记的sPD-L1显著降低了PD-1表达细胞的染色强度,表明该化合物干扰了sPD-L1与细胞表面PD-1的相互作用。 [4] BMS-1001可诱导溶液中PD-L1的二聚化,交联和尺寸排阻色谱法证实了这一点。[4] BMS-1001与PD-L1复合物的晶体结构(PDB 5NIU)显示,该化合物结合于两个PD-L1单体界面处的隧道中,诱导Tyr56侧链旋转,并与Asp122、Lys124形成氢键,以及与Tyr123和Lys124发生水介导的相互作用。[4] |
| 体内研究 (In Vivo) |
NP19(BMS-1类似物)在H22肝癌小鼠模型中的体内抗肿瘤活性[5]
鉴于NP19在黑色素瘤B16-F10肿瘤模型中表现出优异的体内抗肿瘤疗效,且PD-1/PD-L1抑制剂具有广谱抗肿瘤活性,我们进一步利用BALB/c小鼠H22肝癌肿瘤模型评估了化合物NP19的体内抗肿瘤疗效。每只小鼠右侧腹部皮下注射80万个H22细胞。待肿瘤体积达到约100 mm³后,将小鼠随机分组,并分别腹腔注射NP19或载体溶液,持续14天。如图 8 所示,NP19 在 25 mg/kg 剂量下表现出显著的体内抗肿瘤疗效,肿瘤生长抑制率 (TGI) 为 76.5%(图 8A、8B、8C)。此外,NP19 未引起明显的体重下降(图 8D),表明该化合物具有良好的耐受性。 NP19(BMS-1 的类似物)在 B16-F10 小鼠黑色素瘤模型中的体内抗肿瘤活性[5] 为了确定新合成化合物的体外抗 PD-1/PD-L1 活性是否能转化为体内疗效,我们测试了化合物 NP19 在小鼠黑色素瘤 B16-F10 肿瘤模型中的抗肿瘤活性。由于NP19合成简便且细胞毒性较低(表9),因此被选用于体内疗效研究,相比之下,NP2或NP12的活性更高。我们用载体对照组和NP19(25 mg/kg、50 mg/kg、100 mg/kg)对携带黑色素瘤的BALB/c小鼠进行治疗,NP19通过灌胃法每日一次给药,持续15天。如图6所示,治疗15天后,NP19治疗显著抑制了黑色素瘤的生长。 NP19的体内药代动力学特性[BMS-1类似物][5] 由于化合物NP19在体外显示出高活性,接下来通过静脉注射和口服给药,在雄性Sprague-Dawley大鼠中评估了其药代动力学(PK)特征。表8总结了口服和静脉给药的关键药代动力学参数。单次静脉注射1 mg/kg化合物NP19后,NP19的半衰期(t1/2)、清除率(CL)和表观分布容积(Vss)分别为1.5 ± 0.5 h、0.9 ± 0.2 L/h/kg和2.1 ± 0.5 L/kg。口服10 mg/kg NP19后,观察到口服吸收良好(Tmax = 0.6 ± 0.2 h)、半衰期较长(t1/2 = 10.9 ± 7.7 h)和口服生物利用度较高(F = 5%)。此外,在大鼠中未观察到明显的不良反应。与静脉注射半衰期(1.5 h)相比,NP19经灌胃给药后的半衰期(10.9 h)显著延长。这可能是由于NP19的高亲脂性(logP = 7.9)或较差的水溶性所致。因此,NP19表现出翻转药代动力学特征。这种翻转药代动力学特征有时会出现在水溶性差的化合物中,例如瑞巴派特,其t1/2(po)/t1/2(iv)比值为13.5,这是由于其水溶性差(7.6 μg/mL)。另一个例子是李建明等人报道的亲脂性化合物IAT(一种抗微管蛋白药物,水溶性为19 μg/mL),其t1/2(po)/t1/2(iv)比值约为5,与NP19[t1/2(po)/t1/2(iv) = 7.1]相似。由于化合物NP19的口服生物利用度较低,我们推测需要高剂量才能达到足够的药物浓度以发挥抗肿瘤疗效。因此,我们进一步研究了化合物NP19的体内活性。 当PD-1/PD-L1抑制剂1与解毒三元汤(JSD)联合使用时,PI3K/AKT信号通路可显著抑制和逆转上皮-间质转化(EMT)。[3] |
| 酶活实验 |
体外PD-1/PD-L1结合试验[5]
\n采用PD-1/PD-L1均相时间分辨荧光(HTRF)结合试验研究化合物抑制PD-1/PD-L1相互作用的能力。PD-1/PD-L1结合试验试剂盒购自Cisbio公司。实验按照说明书进行,说明书可从https://www.cisbio.com/usa/drug-discovery/human-pd1pd-l1-biochemical-interaction-assay下载。\n \n\n\n百时美施贵宝(BMS)近期公布了首个针对PD-1/PD-L1通路的非肽类小分子抑制剂,该抑制剂在均相时间分辨荧光(HTRF)结合试验中显示出活性;然而,并未提供进一步的数据来支持其活性。 \n\n核磁共振测量[4] \n通过在以15NH4Cl为唯一氮源的M9最小培养基中表达蛋白质,获得均匀的15N标记。进行核磁共振测量时,通过凝胶过滤将缓冲液替换为pH 7.4的PBS缓冲液(PD-L1)或pH 6.4的含100 mM NaCl的25 mM磷酸钠缓冲液(PD-1)。向样品中加入10% (v/v)的D2O以提供锁定信号。所有谱图均使用Bruker Avance III 600 MHz核磁共振波谱仪在300 K下记录。通过监测化合物滴定过程中1H-15N二维HMQC谱中核磁共振化学位移的扰动,评估化合物与PD-L1的相互作用。使用拮抗剂诱导解离试验评估受试化合物解离 PD-L1/PD-1 的能力。简而言之,用未标记的 PD-L1 对 15N 标记的 PD-1 (0.2 mM) 进行略微过量滴定。将化合物分装到所得混合物中。实验过程中,使用 HMQC 监测 1H-15N 信号。\n \n\nPD1/PD-L1 检查点检测[4] \n在检测前 24 小时,将 aAPC 以每孔 10,000 个细胞的密度接种于白色 96 孔板中,并置于培养基中。在检测当天,将抗体在含 1% FBS 的 RPMI 1640 培养基中进行 3.5 倍系列稀释。将BMS化合物[BMS-1]用DMSO进行系列稀释,并配制成含1% FBS的RPMI 1640培养基。这样可以确保所有样品中DMSO的浓度恒定。从孔中移除95 μl培养基,并在细胞上覆盖40 μl化合物稀释液。向每个孔中加入40 μl含1% FBS的RPMI 1640培养基,其中包含20,000个内皮细胞(EC)。在37°C孵育6小时后,将培养板在室温下平衡10分钟,然后向每个孔中加入80 μl Bio-Glo试剂。孵育 10 分钟后,使用 FlexStation 3 对发光强度进行定量。通过将 Hill 方程拟合到实验数据来确定半数最大有效浓度 (EC50) 和最大发光值 (RLUmax)。 \nPD-1/sPD-L1 效应细胞测定[4] \n为了评估 BMS 对可溶性 PD-L1 抑制 T 细胞的影响,在重组人 sPD-L1 存在的情况下,用抗 CD3 抗体刺激内皮细胞 (EC)。为此,将 96 孔白色平底板在 4°C 下用 5 μg/ml 的抗 CD3 抗体或同型对照抗体 PBS 溶液包被过夜。去除抗体溶液,用 PBS 洗涤孔板 3 次并干燥。将 sPD-L1(氨基酸 18–134)溶于添加了青霉素/链霉素溶液(终浓度均为 100 U/ml)的 PBS 缓冲液中,并加入 BMS 化合物 [BMS-1] 或等体积的 DMSO。然后,将 15 μl 该溶液加入到抗体包被板的每个孔中。离心收集内皮细胞 (EC) 并稀释至 50,000 个细胞/ml,取 60 μl 细胞悬液加入到每个孔中。sPD-L1 的终浓度为 10 μg/ml (0.6 μM)。BMS 化合物 [BMS-1] 的终浓度分别为:0.12、0.3、1.2 和 3 μM,对应的 BMS 与 sPD-L1 的摩尔比分别为:1:5、1:2、2:1 和 5:1。细胞培养24小时后,使用Bio-Glo荧光素酶检测试剂盒,按照制造商说明书进行荧光素酶活性测定。 通过监测1H-15N二维HMQC谱中NMR共振化学位移的变化,评估BMS-1001与PD-L1的结合情况。使用均匀15N标记的PD-L1,并用化合物进行滴定。线宽展宽表明化合物诱导形成了更高分子量的复合物。[4] 使用拮抗剂诱导解离试验(AIDA)评估BMS-1001解离预先形成的PD-1/PD-L1复合物的能力。用未标记的PD-L1滴定15N标记的PD-1形成复合物,然后加入化合物。 PD-1窄1H-15N信号的恢复表明复合物被破坏。[4] hPD-L1/BMS-1001复合物的结晶采用悬滴气相扩散法,在室温下进行,结晶溶剂为0.1 M Tris pH 8.5,含有0.2 M氯化镁和30% (w/v) PEG 4000。X射线衍射数据在PETRA III P11光束线收集,结构通过分子置换法解析,以PDB 5C3T为搜索模型,精修至2.01 Å分辨率。[4] |
| 细胞实验 |
MTS 法用于测定细胞增殖。在 96 孔板的每个孔中,将 1-2×10³ 个细胞接种于 100 μl 培养基中。24 小时后,在测试培养基(含 1% 胎牛血清的 DMEM)中加入 PD-1/PD-L1 抑制剂 1 和 ERK1/2 抑制剂,单独或联合处理,每个处理设置三个复孔,持续 72 小时,浓度需稀释至所需水平。
细胞系[4] 为了验证 BMS 化合物 [BMS-1] 抑制 PD-1/PD-L1 相互作用的效力,我们使用了基于细胞的 PD-1/PD-L1 免疫检查点阻断模型。本实验使用了两种模型细胞系:人工抗原呈递细胞(PD-L1+ aAPC/CHO-K1细胞,简称aAPC),其过表达TCR配体和PD-L1;以及T细胞替代物,即过表达PD-1并携带NFAT启动子控制的荧光素酶报告基因的改良Jurkat T细胞系(PD-1效应细胞,简称EC)。这些细胞购自Promega公司,并在添加了10%胎牛血清、100 U/ml青霉素和100 U/ml链霉素的RPMI 1640培养基中培养。此外,为了确保导入的基因构建体的稳定表达,细胞培养过程中持续加入潮霉素B(50 μg/ml)和G418(250 μg/ml)。在实验中,后两种抗生素被省略。通过流式细胞术验证了内皮细胞 (EC) 上 PD-1 的过表达和抗原呈递细胞 (aAPC) 上 PD-L1 的过表达(数据未显示),并通过监测抗 CD3 抗体刺激后的荧光素酶活性验证了荧光素酶表达基因的存在。抗生素筛选、流式细胞术和报告基因表达作为细胞系鉴定方法。定期检测细胞,并使用基于 PCR 的方法检测支原体污染,结果均为阴性。 细胞毒性试验[4] 将 5000 个内皮细胞接种于透明的 96 孔板中,并在浓度递增的 BMS 化合物 [BMS-1] 或 DMSO(作为对照,所有样品中 DMSO 的浓度均保持不变)存在下培养 48 小时。治疗后,根据制造商的说明,使用 Biolog Redox Dye Mix MB 进行代谢活性检测。 流式细胞术测量[4] 通过流式细胞术评估 sPD-L1(氨基酸 18–134)与内皮细胞 (EC) 的结合。将 His 标签的 PD-L1 蛋白或其突变体与 NTA-Atto 647 N 荧光染料在 22°C 下以 8:1 的摩尔比(蛋白:染料)染色 2 小时。将 PD-L1-Atto 与待测化合物或抗体配制在 150 μl PBS 中。样品在 4°C 避光孵育 30 分钟。同时,离心内皮细胞,用 PBS 洗涤,并重悬于新鲜 PBS 中,浓度为 1 × 10⁶ 个细胞/ml。向每个样品中加入 50 μl 内皮细胞悬液,并在冰上孵育 60 分钟。各组分的最终浓度分别为:PD-L1 25 μg/ml (1.5 μM)、抗PD-L1抗体和对照抗体125 μg/ml,以及BMS化合物[BMS-1] 1 μM。使用BD FACS Verse流式细胞仪和BD FACSuite v1.0.6软件分析样品。 采用代谢活性测定法评估BMS-1001的细胞毒性。将修饰的Jurkat T细胞(PD-1效应细胞)暴露于递增浓度的化合物中48小时,并使用Biolog Redox Dye Mix MB测定代谢活性。细胞毒性的EC50值为33.4 μM。 [4] PD-1/PD-L1 检查点检测:将 aAPC(过表达 TCR 配体和 PD-L1 的 CHO-K1 细胞)接种于 96 孔板中。用 DMSO 对 BMS-1001 进行系列稀释,并用含 1% FBS 的 RPMI 1640 培养基配制。加入 PD-1 效应细胞(表达 PD-1 和 NFAT-荧光素酶的 Jurkat T 细胞)。孵育 6 小时后,加入 Bio-Glo 试剂并定量检测发光值。该化合物呈剂量依赖性地诱导荧光素酶活性。[4] sPD-L1 效应细胞检测:用抗 CD3 抗体包被 96 孔板。将 sPD-L1(氨基酸 18-134)用含青霉素/链霉素的 PBS 稀释,并加入 BMS-1001 或 DMSO,然后加入包被的培养板,再加入 PD-1 效应细胞。sPD-L1 的最终浓度为 10 μg/mL (0.6 μM),BMS-1001 的浓度分别为 0.12、0.3、1.2 和 3 μM(摩尔比分别为 1:5、1:2、2:1 和 5:1)。24 小时后,测定荧光素酶活性。[4] 流式细胞术结合实验:用 NTA-Atto 647 N 荧光染料标记 His 标签的 sPD-L1。将标记的 sPD-L1 与 BMS-1001 (1 μM) 或抗体在 4°C 下预孵育 30 分钟,然后加入 PD-1 效应细胞,并在冰上孵育 60 分钟。通过流式细胞术分析细胞染色。[4] |
| 动物实验 |
雄性Sprague-Dawley大鼠药代动力学研究[5]
本研究采用雄性Sprague-Dawley大鼠(200-220 g)研究化合物NP19(BMS-1类似物)的药代动力学。实验前12小时禁食,但可自由饮水。分别于口服(10 mg/kg)或静脉注射(1 mg/kg)化合物NP19后0.0833、0.25、0.5、1、1.5、2、4、6、8、12和24小时,从尾静脉采集血样(0.3 mL)至肝素化1.5 mL聚乙烯管中。该化合物溶于 5% DMSO 和 95% PEG-300 的混合溶液中用于静脉注射,或悬浮于 0.5% 羧甲基纤维素钠 (CMC-Na) 溶液中用于口服。样品立即以 3000 g 离心 10 分钟。所得血浆 (100 μL) 储存于 -20 °C 直至分析。使用 DAS(药物与统计)软件,通过非房室模型分析,根据个体动物数据确定药代动力学 (PK) 参数。PK 研究的仪器和分析条件:使用配备电喷雾电离 (ESI) 接口的 ACQUITY I-Class UPLC 和 XEVO TQD 三重四极杆质谱仪(Waters 公司,美国马萨诸塞州米尔福德)的 UPLC-MS/MS 系统分析血液样本。UPLC 系统由二元溶剂管理器 (BSM) 和带流通针的样品管理器 (SM-FTN) 组成。使用 Masslynx 4.1 软件(Waters 公司)进行数据采集和仪器控制。采用多反应监测 (MRM) 模式,NP19 的 m/z 555.35 → 181.03 和卡马西平的 m/z 237 → 194.1 进行定量分析。 小鼠 B16F10 黑色素瘤模型体内疗效研究[5] 使用 6-8 周龄的 BALB/c 小鼠研究 NP19(BMS-1 类似物)对皮下移植黑色素瘤细胞模型的抑制作用。将处于对数生长期的小鼠 B16F10 黑色素瘤细胞悬浮于 PBS 中,浓度为 2 × 10⁶ 个/mL。每只小鼠皮下接种 200 μL 含有 4 × 10⁵ 个细胞的细胞悬液。当肿瘤体积达到约100 mm³时,将小鼠随机分为四组(n = 10),分别给予NP19(25、50、100 mg/kg)和溶剂对照。药物通过灌胃法每日一次给药,持续15天。溶剂对照组灌胃0.5%羧甲基纤维素钠(CMC-Na)。在整个实验期间监测动物的活动和体重,以评估急性毒性。治疗开始16天后处死小鼠,收集肿瘤组织和主要器官(肝脏、脾脏、胸腺和肾脏)样本。收集的肿瘤组织和器官(肝脏、肾脏)用4%多聚甲醛固定,常规石蜡包埋,苏木精-伊红(H&E)染色,并在显微镜下观察。肿瘤生长抑制值 (TGI) 的计算公式为:TGI(%) = [1 – Wt/Wv] × 100%,其中 Wt 和 Wv 分别为治疗组和载体对照组的平均肿瘤重量。 小鼠 H22 肝癌模型体内疗效研究[5] 使用 6-8 周龄雄性 BALB/c 小鼠。根据肿瘤移植研究方案,将 8 × 10⁵ 个 H22 细胞接种到每只小鼠的右侧腹部。NP19(BMS-1 的类似物)溶解于 5% DMSO、40% PEG-200 和 55% 生理盐水中,配制成所需浓度。对照组小鼠腹腔注射 200 μL 载体溶液。每隔2天使用可溯源的电子数显卡尺测量肿瘤体积,并使用公式a × b² × 0.5计算,其中a和b分别代表肿瘤的长径和短径。治疗结束后处死小鼠,切除肿瘤并称重。 |
| 毒性/毒理 (Toxicokinetics/TK) |
BMS-1001在代谢活性测定中对Jurkat T细胞表现出低毒性,处理48小时后EC50为33.4 μM。[4]
使用修饰的CHO-K1细胞(aAPCs)也证实了其低毒性。[4] 其他BMS化合物(BMS-37和BMS-242)的毒性更高,EC50介于3-6 μM之间,而BMS-1001的毒性则显著降低。[4] |
| 参考文献 | |
| 其他信息 |
目的:三阴性乳腺癌(TNBC)预后不良,除化疗外,缺乏有效的全身治疗手段。靶向免疫检查点PD-1/PD-L1在乳腺癌,尤其是TNBC的治疗中显示出良好的治疗前景。细胞外信号调节激酶1/2(ERK1/2)是肿瘤发生的重要驱动因素。本研究旨在探讨PD-1/PD-L1抑制剂与ERK1/2抑制剂联合治疗对乳腺癌细胞系生长和细胞内功能的影响。方法:在三种不同的TNBC细胞亚型和一种非TNBC细胞系中测定各抑制剂的IC50值以及联合治疗的疗效。采用磷酸化特异性抗体进行Western blot分析,以研究其对ERK1/2激活的影响。采用定量逆转录PCR检测免疫调节基因和细胞周期相关基因的表达。结果:PD-1/PD-L1和ERK1/2抑制剂均能更有效地抑制三阴性乳腺癌(TNBC)细胞的增殖,且抑制程度高于非TNBC细胞。联合用药可对细胞系产生协同或叠加的抑制作用。ERK1/2抑制剂,无论单独使用还是联合使用,均可降低ERK1/2和S6的磷酸化水平,并降低c-Fos和FosL的表达。在免疫调节相关基因中,联合治疗后TNBC细胞中IL-8的表达上调。结论:PD-1/PD-L1和ERK1/2抑制剂的联合应用显示出良好的治疗效果,有望成为一种新的治疗策略,其在TNBC细胞系中的疗效优于非TNBC细胞。这些效果需要在能够反映免疫细胞与肿瘤细胞相互作用的模型(例如肿瘤微环境)中进行验证。 [2]
肠道菌群、食物摄入和胆汁在吲哚美辛诱导的大鼠小肠炎症中发挥重要作用。牛磺熊去氧胆酸 (TUDCA) 和熊去氧胆酸 (UDCA) 可抑制疏水性胆汁酸诱导的多种细胞损伤。我们研究了这些胆汁酸以及其他胆汁酸对该炎症模型的影响。在给予大鼠为期 7 天的胆汁酸预处理后,注射吲哚美辛,我们评估了临床和肠道炎症指标以及胆汁分泌情况。UDCA 显著增强了吲哚美辛诱导的食物摄入量和体重下降、总体炎症评分和髓过氧化物酶活性升高以及小肠长度缩短。牛磺酸鹅脱氧胆酸 (TCDCA) 显著改善了临床炎症指标,抑制了吲哚美辛诱导的胆汁中次级胆汁酸含量和疏水性指数的升高,并有减轻肠道炎症的趋势。尽管在 TCDCA 组小鼠注射吲哚美辛前,胆汁酸水平和胆汁疏水性指数分别出现升高和降低,但这些变化并不能解释 TCDCA 介导的保护作用。膳食 TCDCA 可减轻模型中的肠道炎症,而熊去氧胆酸 (UDCA) 则会加剧炎症。胆汁成分的变化(UDCA 和鹅脱氧胆酸的增加)可能解释了观察到的调节作用。[3] BMS-1001 是百时美施贵宝公司专利(WO2015034820、WO2015160641)中优化的小分子抑制剂之一。它与PD-L1结合并诱导PD-L1二聚化,从而使PD-L1两个亚基的PD-1结合表面均能被PD-L1亚基所占据。晶体结构显示,2,3-二氢-1,4-苯并二恶英部分诱导Tyr56侧链的运动,将结合口袋转化为一个隧道。(2R)-2-氨基-3-羟基丙酸部分与Asp122羰基之间形成氢键,并通过水分子介导与Lys124 NH2和Tyr123主链羰基形成氢键。3-氰基苄基取代基与Tyr123和Arg125形成疏水相互作用。[4] |
| 分子式 |
C29H33NO5
|
|
|---|---|---|
| 分子量 |
475.58
|
|
| 精确质量 |
475.235
|
|
| 元素分析 |
C, 73.24; H, 6.99; N, 2.95; O, 16.82
|
|
| CAS号 |
1675201-83-8
|
|
| 相关CAS号 |
|
|
| PubChem CID |
91663303
|
|
| 外观&性状 |
White to off-white crystalline solid
|
|
| 密度 |
1.2±0.1 g/cm3
|
|
| 沸点 |
630.2±55.0 °C at 760 mmHg
|
|
| 闪点 |
334.9±31.5 °C
|
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
|
| 折射率 |
1.590
|
|
| LogP |
5.18
|
|
| tPSA |
68.2
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
9
|
|
| 重原子数目 |
35
|
|
| 分子复杂度/Complexity |
639
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
O([H])C([C@]1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])N1C([H])([H])C1C(=C([H])C(=C([H])C=1OC([H])([H])[H])OC([H])([H])C1C([H])=C([H])C([H])=C(C2C([H])=C([H])C([H])=C([H])C=2[H])C=1C([H])([H])[H])OC([H])([H])[H])=O
|
|
| InChi Key |
ZBOYJODMIAUJHH-SANMLTNESA-N
|
|
| InChi Code |
InChI=1S/C29H33NO5/c1-20-22(12-9-13-24(20)21-10-5-4-6-11-21)19-35-23-16-27(33-2)25(28(17-23)34-3)18-30-15-8-7-14-26(30)29(31)32/h4-6,9-13,16-17,26H,7-8,14-15,18-19H2,1-3H3,(H,31,32)/t26-/m0/s1
|
|
| 化学名 |
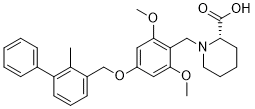
(2S)-1-[[2,6-dimethoxy-4-[(2-methyl-3-phenylphenyl)methoxy]phenyl]methyl]piperidine-2-carboxylic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1 mg/mL (2.10 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 10.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1 mg/mL (2.10 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 10.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1 mg/mL (2.10 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 10 mg/mL (21.03 mM) in 50% PEG300 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1027 mL | 10.5135 mL | 21.0270 mL | |
| 5 mM | 0.4205 mL | 2.1027 mL | 4.2054 mL | |
| 10 mM | 0.2103 mL | 1.0513 mL | 2.1027 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
