| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
MEK1; MEK2
Mitogen-activated protein kinase kinase 1 (MEK1) and MEK2, serine/threonine kinases in the MAPK pathway. For Pimasertib (SAR245509, AS703026, MSC1936369B), the IC50 values from [1] were: MEK1 = 7 nM, MEK2 = 15 nM (HTRF kinase assay). It showed no inhibition of 29 other kinases (e.g., ERK1, JNK, p38, PI3K) at 1 μM, confirming MEK1/2 selectivity [1] - No new target potency data; focus on overcoming BRAF inhibitor resistance without additional MEK parameters [2] - Target consistent with [1], no additional numerical data (focus on KRAS-mutant colorectal cancer) [3] |
|---|---|
| 体外研究 (In Vitro) |
Pimasertib(5、0.5 和 0.1 μM)特异性抑制单独培养或与 BMSC 一起培养的 MM 细胞中的 ERK1/2 激活。 Pimasertib 的 IC50 值范围为 0.005 至 2 M,表明它以剂量依赖性方式抑制 MM 细胞系的生长。 Pimasertib 对于 INA-6、U266 和 H929 细胞的 IC50 值分别为 10 nM、5 nM 和 200 nM。 pimasertib 可以改变细胞凋亡和细胞周期特征。 BM微环境是pimasertib对MM细胞作用的目标[1]。在对西妥昔单抗耐药的 D-MUT 细胞中,pimasertib (10 mol/L) 抑制 ERK 通路、增殖和转化[2]。与单独使用每种药物相比,pimasertib 与 PLX4032 联合使用时显着增加 RPMI-7951 细胞发生凋亡的可能性。为了达到与 PLX4032 和 Pimasertib 联合治疗相当的结果,Pimasertib 与小干扰 RNA 介导的 BRAF 下调协同作用[3]。
多发性骨髓瘤(MM)细胞:在MM细胞系(RPMI-8226、U266、5T33MM)中,Pimasertib(0.01 μM–10 μM)抑制增殖,MTT法(72小时)测得IC50分别为RPMI-8226 0.12 μM、U266 0.18 μM、5T33MM 0.2 μM。Western blot显示RPMI-8226细胞经0.5 μM处理2小时后p-ERK减少85%;Annexin V-FITC染色显示1 μM处理48小时后凋亡率达45%。此外,该药还可减少IL-6诱导的STAT3磷酸化(0.5 μM时减少60%) [1] - BRAF突变黑色素瘤细胞:在对PLX4032(BRAF抑制剂)耐药的A375细胞(A375-R)中,Pimasertib(0.05 μM–5 μM)恢复敏感性:IC50=0.2 μM(A375-R),亲本A375细胞为0.15 μM(CCK-8法,72小时)。Western blot显示A375-R细胞中1 μM处理后p-ERK减少90%、p-MEK减少80% [2] - KRAS突变结直肠癌细胞(CRC):在对EGFR单抗(西妥昔单抗)耐药的KRAS突变CRC细胞(HCT116、SW480)中,Pimasertib(0.1 μM–10 μM)抑制增殖,MTT法(72小时)测得IC50为HCT116 0.3 μM、SW480 0.4 μM。该药可减少cyclin D1(1 μM时qRT-PCR检测减少55%),并增强西妥昔单抗诱导的凋亡(单独用药凋亡率20%,联合0.5 μM Pimasertib后达50%) [3] |
| 体内研究 (In Vivo) |
Pimasertib (15、30 mg/kg) 显着减缓携带人 H929 MM 异种移植物的 CB17 SCID 小鼠的肿瘤生长[1]。 Pimasertib(10 mg/kg,口服)可抑制因 K-ras 基因突变而对西妥昔单抗产生耐药性的肿瘤的生长[2]。
多发性骨髓瘤小鼠模型:5T33MM同系小鼠(n=10/组)随机分为溶媒组(0.5%甲基纤维素+0.1%吐温80)和Pimasertib 20 mg/kg组。药物口服每日一次,连续28天。较溶媒组,肿瘤负荷(血清M蛋白)减少60%,生存期延长40%(中位生存期:42天 vs. 30天) [1] - 黑色素瘤异种移植模型:6周龄雌性裸鼠接种A375-R细胞,用Pimasertib 15 mg/kg(口服每日一次)处理21天。肿瘤体积较溶媒组减少55%,肿瘤组织中p-ERK(Western blot)减少75% [2] - 结直肠癌异种移植模型:7周龄雄性裸鼠接种HCT116细胞,用Pimasertib 25 mg/kg(口服每日一次)±西妥昔单抗10 mg/kg(腹腔注射每周两次)处理28天。肿瘤体积减少率:单独Pimasertib组50%、单独西妥昔单抗组30%、联合组75%。血清CEA从500 ng/mL降至150 ng/mL(联合组) [3] |
| 酶活实验 |
AS703026 溶解在 2.5% DMSO 中。激活的二磷酸化 MEK (pp-MEK) 检测包含 40 M 33P-γATP (AppKm 8.5 MμM、0.5 nM 人激活 MEK1 或 MEK2 和 1 M 激酶死亡 ERK2 (AppKm 0.73 μM)。所有测试均在含有以下成分的缓冲液中进行: 20 mM HEPES (pH 7.2)、5 mM 2-巯基乙醇、0.15 mg/mL BSA 和 10 mM MgCl2。对于所有测定,最终 33P-ATP 浓度为 0.02 μCi/μL。40 分钟后,pp-MEK 激酶反应通过将 30 μL 反应混合物转移至含有 12.5% TCA 的 Durapore 0.45-μm 过滤板来停止。将过滤器干燥,然后使用液体闪烁剂在 TopCount 上读取。对于 IC50,检查浓度响应数据。最初未磷酸化的 MEK 的 IC50 (u-MEK) 的计算方法是将 0.2 nM 重组人 MEK1 或 MEK2 与载体或 AS703026 在反应缓冲液中预孵育 40 分钟。通过添加最终浓度 20 nM B-RafV600E 和 30 μM ATP 10 分钟,磷酸化/激活然后加入B-Raf抑制剂SB590885(终浓度100 nM),猝灭B-Raf活性,并通过在反应缓冲液中添加1 μM KD-ERK2和0.02 μCi/μL 33P-ATP来测量MEK激酶活性。将 30μL 反应混合物转移至 Durapore 滤板并照常读数,90 分钟后激酶反应停止。
MEK1/2 HTRF激酶实验:将重组人MEK1(44–313位氨基酸)或MEK2(38–326位氨基酸)与生物素化肽底物(MEK1:RRRVSYRRR,MEK2:RRRLSYRRR,20 μM)、Eu标记抗磷酸肽抗体及ATP(10 μM)共同孵育于激酶缓冲液(25 mM Tris-HCl pH 7.5、10 mM MgCl₂、1 mM DTT)中。加入系列稀释的Pimasertib(0.001 nM–100 nM),30°C孵育60分钟。检测时间分辨荧光(激发光340 nm,发射光620 nm),通过四参数逻辑回归计算IC50 [1] |
| 细胞实验 |
[3H]胸苷掺入和MTT染料吸光度测量均用于确定研究化合物对MM细胞生长和存活的抑制作用。在 96 孔板中,细胞以每孔 104 个细胞的密度一式三份以及每孔 2-5×105 个细胞的密度培养 3 天(MM 细胞系)或 5 天(患者 MM 细胞)。对于 [3H] 胸苷掺入测定,细胞用 0.5 μCi (0.0185 MBq)/孔 [3H]胸苷脉冲 6 小时(细胞系),收获到玻璃纤维过滤器上,并在 β-闪烁计数器中计数。由于患者的 MM 细胞的 DNA 合成水平较低,因此用 2 μCi/孔的 [3H]胸苷对其进行脉冲,并在培养的最后 36 小时内测量其 DNA 合成。
MM细胞增殖与凋亡实验:RPMI-8226/U266细胞以5×10³个细胞/孔接种于96孔板,用Pimasertib(0.01 μM–10 μM)处理72小时。加入5 mg/mL MTT试剂孵育4小时,DMSO溶解甲臜结晶后,检测570 nm吸光度计算IC50。凋亡实验中,细胞(2×10⁵个/孔,6孔板)用1 μM Pimasertib处理48小时,Annexin V-FITC/PI染色,流式细胞术分析 [1] - PLX4032耐药黑色素瘤实验:A375-R细胞以5×10³个细胞/孔接种于96孔板,用Pimasertib(0.05 μM–5 μM)处理72小时,CCK-8试剂检测活力。Western blot实验中,细胞(3×10⁵个/孔,6孔板)用1 μM Pimasertib处理2小时,RIPA缓冲液裂解后,用抗p-ERK、抗p-MEK及抗GAPDH抗体检测 [2] - CRC细胞协同实验:HCT116细胞以5×10³个细胞/孔接种于96孔板,用Pimasertib(0.1 μM–10 μM)±西妥昔单抗(10 μg/mL)处理72小时,MTT法检测增殖;Annexin V染色分析凋亡。qRT-PCR实验中,细胞用1 μM Pimasertib处理24小时,定量cyclin D1 mRNA [3] |
| 动物实验 |
将 H929 细胞(4×10⁶ 个)皮下注射到 CB17 重症联合免疫缺陷 (SCID) 小鼠体内,注射液为 100 μL RPMI-1640 培养基。注射后第三周,小鼠出现可触及的肿瘤(约 130 mm³),之后每天两次口服给予 Pimasertib(15 或 30 mg/kg)或对照溶剂。每隔一天,使用游标卡尺测量肿瘤的二维尺寸,并计算肿瘤体积。当动物的生活质量显著下降,肿瘤体积增大至 2 cm³,或出现濒死状态,或出现瘫痪时,即出现上述情况。使用 GraphPad Prism 4.03 for Windows 软件绘制对照药物组和 Pimasertib 组小鼠肿瘤形成情况的变化曲线。利用特异性单克隆抗体 (m) 对肿瘤进行免疫印迹和免疫化学分析。使用Leica IM50图像管理器拍摄图像,使用Leica DM LB研究显微镜进行图像分析,并使用Adobe Photoshop 7.0软件进行后期处理。
5T33MM同源模型方案:将8周龄的5T33MM小鼠通过灌胃给予Pimasertib(20 mg/kg,溶于0.5%甲基纤维素+0.1% Tween 80溶液),每日一次,持续28天。对照组小鼠接受相同溶剂。每周通过ELISA检测血清M蛋白水平;每日监测小鼠存活率[1]。 - A375-R黑色素瘤异种移植方案:将5×10⁶个A375-R细胞皮下植入6周龄雌性裸鼠体内。当肿瘤体积达到约100 mm³时,每日一次灌胃给予Pimasertib(15 mg/kg,溶于0.5%甲基纤维素溶液),持续21天。每3天测量一次肿瘤体积(长×宽²/2);切除肿瘤进行p-ERK蛋白质印迹分析[2] - HCT116 CRC异种移植方案:将4×10⁶个HCT116细胞皮下植入7周龄雄性裸鼠体内。当肿瘤体积达到约120 mm³时,小鼠接受Pimasertib(25 mg/kg,口服,每日一次)±西妥昔单抗(10 mg/kg,腹腔注射,每周两次)治疗,疗程28天。每周通过ELISA检测血清CEA水平;每3天记录一次肿瘤体积[3] |
| 药代性质 (ADME/PK) |
在雄性Sprague-Dawley大鼠中,口服Pimasertib(20 mg/kg)的口服生物利用度为52%,Cmax = 3.5 μM,Tmax = 1.2小时,末端半衰期为6.8小时[1]
- 大鼠静脉注射Pimasertib(5 mg/kg)的清除率(CL)为8.3 mL/min/kg,稳态分布容积(Vss)为1.1 L/kg[1] - 通过平衡透析法测得Pimasertib的人血浆蛋白结合率为97%[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
体外细胞毒性:在正常人外周血单核细胞 (PBMC) 和包皮成纤维细胞中,Pimasertib(浓度高达 10 μM,处理 72 小时)的细胞活力 > 85%,表明其非特异性毒性较低 [1][2][3]
- 体内急性毒性:在接受 Pimasertib(20 mg/kg,口服,28 天)治疗的大鼠中,未观察到明显的体重减轻、嗜睡或血清 ALT/AST/肌酐水平异常。肝脏/肾脏组织学检查未见炎症或坏死 [1] |
| 参考文献 |
|
| 其他信息 |
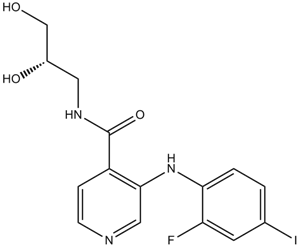
N-[(2S)-2,3-二羟丙基]-3-(2-氟-4-碘苯胺基)-4-吡啶甲酰胺是一种吡啶甲酰胺类化合物。
Pimasertib 正在进行临床试验 NCT01378377(Pimasertib (MSC1936369B) 与 Temsirolimus 联合用药试验)。 Pimasertib 是一种口服生物利用度高的小分子 MEK1 和 MEK2 (MEK1/2) 抑制剂,具有潜在的抗肿瘤活性。Pimasertib 选择性地结合并抑制 MEK1/2 的活性,从而阻止 MEK1/2 依赖性效应蛋白和转录因子的激活,这可能导致生长因子介导的细胞信号传导和肿瘤细胞增殖受到抑制。 MEK1/2 (MAP2K1/K2) 是双特异性苏氨酸/酪氨酸激酶,在 RAS/RAF/MEK/ERK 信号通路的激活中发挥关键作用,并在多种肿瘤细胞类型中常呈高表达。我们研究了一种新型、选择性、口服生物利用度高的 MEK1/2 抑制剂 AS703026 在人多发性骨髓瘤 (MM) 中的细胞毒性和作用机制。AS703026 抑制 MM 细胞生长和存活以及细胞因子诱导的破骨细胞分化的能力比 AZD6244 强 9-10 倍。AS703026 诱导的增殖抑制是通过 G0/G1 期细胞周期阻滞介导的,并伴有 c-maf 癌基因表达的降低。 AS703026 可进一步通过 caspase 3 和 PARP 裂解诱导多发性骨髓瘤 (MM) 细胞凋亡,无论是否存在骨髓基质细胞 (BMSCs)。重要的是,AS703026 可增强 MM 细胞对多种传统抗 MM 药物(地塞米松、美法仑)以及新型或新兴抗 MM 药物(来那度胺、培利福辛、硼替佐米、雷帕霉素)的敏感性。在携带 H929 MM 异种移植瘤的小鼠中,AS703026 治疗组与载体对照组相比,肿瘤生长显著减少,这与 pERK1/2 下调、PARP 裂解诱导以及体内微血管减少相关。此外,AS703026 (<200 nM) 对大多数复发/难治性 MM 患者的肿瘤细胞 (84%) 具有细胞毒性,且与 RAS 和 BRAF 基因的突变状态无关。重要的是,在相同的剂量范围内,BMSC诱导的MM患者细胞活力同样被抑制。因此,我们的结果支持对AS703026进行临床评估,无论单独使用还是与其他抗MM药物联合使用,以改善患者的预后。[1] 背景:尽管原癌基因BRAF抑制剂已显示出对恶性黑色素瘤的优异抗肿瘤活性,但其疗效受限于获得性耐药性的产生,而MAP激酶(MEK)的重新激活在这一过程中发挥着重要作用。在本研究中,我们评估了新型MEK抑制剂AS703026在BRAF抑制剂耐药的黑色素瘤细胞系中的疗效。方法:我们用BRAF抑制剂PLX4032处理两种携带BRAF激活突变(V600E)的黑色素瘤细胞系RPMI-7951和SK-MEL5,以筛选出BRAF抑制剂耐药细胞系用于进一步研究。采用MTS [3-(4,5-二甲基噻唑-2-基)-5-(3-羧基甲氧基苯基)-2-(4-磺基苯基)-2H-四唑] 法和台盼蓝排除法测定细胞活力;采用Annexin-V染色法进行细胞凋亡检测。采用小干扰RNA(siRNA)技术研究BRAF基因的敲低情况。结果:与单独使用PLX4032或AS703026相比,RPMI-7951细胞对二者联合治疗表现出更高的敏感性。与此一致的是,PLX4032和AS703026联合用药显著诱导细胞凋亡,而单独使用任一药物均未观察到此现象,这通过Annexin-V/碘化丙啶双染细胞的流式细胞术分析和cleaved caspase-3的Western blot分析得到证实。值得注意的是,免疫印迹分析也显示,联合用药治疗可降低磷酸化ERK的水平。此外,AS703026与小干扰RNA介导的BRAF下调具有协同作用,其结果与PLX4032和AS703026联合治疗的结果相似。结论:我们的结果表明,AS703026与BRAF抑制剂联合治疗可克服携带BRAF突变体的恶性黑色素瘤细胞对BRAF抑制剂的耐药性。[2] 表皮生长因子受体(EGFR)单克隆抗体(mAb)广泛用于治疗转移性结直肠癌(mCRC)患者,但目前已明确,携带K-ras突变的患者对EGFR mAb(如西妥昔单抗(爱必妥)和帕尼单抗(维克替比))耐药。因此,目前针对患者的治疗建议包括在接受EGFR单克隆抗体治疗前诊断患者的K-ras突变状态。本研究旨在探讨两种目前正在进行临床试验的MEK抑制剂AS703026和AZD6244能否解决K-ras突变型结直肠癌对EGFR单克隆抗体的耐药性问题。我们利用多种细胞实验和肿瘤异种移植模型对AS703026和AZD6244进行了测试,重点研究了仅表达野生型(WT)或突变型K-Ras(D-WT或D-MUT)的同源人结直肠癌细胞系。EGFR单克隆抗体西妥昔单抗在体外和体内均能抑制Ras-ERK通路和D-WT细胞的增殖,但在任何情况下均不能抑制D-MUT细胞的增殖。相比之下,AS703026 和 AZD6244 通过特异性抑制关键的 MEK 下游靶激酶 ERK,有效抑制了 D-MUT 细胞在体外和体内的生长。AS703026 或 AZD6244 对 MEK 的抑制作用也抑制了由 K-ras 突变引起的西妥昔单抗耐药性结直肠癌细胞在体外和体内的生长。我们的研究结果为MEK抑制剂作为K-ras突变型CRC的有效疗法提供了概念验证。[3] Pimasertib(SAR245509、AS703026、MSC1936369B)是一种选择性口服MEK1/2抑制剂,最初开发用于治疗血液系统恶性肿瘤(例如多发性骨髓瘤)和实体瘤(例如BRAF耐药性黑色素瘤、KRAS突变型结直肠癌)[1][2][3] - 其作用机制包括与MEK1/2的变构位点结合(非ATP竞争性),稳定其非活性构象并阻断ERK磷酸化,从而抑制细胞增殖并诱导细胞凋亡[1][2][3] - 它在两种情况下克服耐药性:通过重新阻断MAPK通路克服黑色素瘤中PLX4032(BRAF抑制剂)耐药性[2],以及克服西妥昔单抗耐药性。通过靶向MEK依赖性生存信号,在KRAS突变型CRC中产生EGFR单抗耐药性[3] |
| 分子式 |
C15H15FIN3O3
|
|---|---|
| 分子量 |
431.20
|
| 精确质量 |
431.014
|
| 元素分析 |
C, 41.78; H, 3.51; F, 4.41; I, 29.43; N, 9.74; O, 11.13
|
| CAS号 |
1236699-92-5
|
| 相关CAS号 |
1236361-78-6 (HCl); 1236699-92-5;
|
| PubChem CID |
44187362
|
| 外观&性状 |
Light yellow to khaki solid powder
|
| 密度 |
1.8±0.1 g/cm3
|
| 沸点 |
623.2±55.0 °C at 760 mmHg
|
| 闪点 |
330.7±31.5 °C
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
| 折射率 |
1.684
|
| LogP |
3.05
|
| tPSA |
94.48
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
391
|
| 定义原子立体中心数目 |
1
|
| SMILES |
FC1=C(C=CC(I)=C1)NC2=CN=CC=C2C(NC[C@@H](CO)O)=O
|
| InChi Key |
VIUAUNHCRHHYNE-JTQLQIEISA-N
|
| InChi Code |
InChI=1S/C15H15FIN3O3/c16-12-5-9(17)1-2-13(12)20-14-7-18-4-3-11(14)15(23)19-6-10(22)8-21/h1-5,7,10,20-22H,6,8H2,(H,19,23)/t10-/m0/s1
|
| 化学名 |
N-[(2S)-2,3-dihydroxypropyl]-3-(2-fluoro-4-iodoanilino)pyridine-4-carboxamide
|
| 别名 |
MSC 1936369B; SAR 245509; AS-703026; SAR245509; SAR-245509; AS703026; AS 703026
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.80 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.80 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.80 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 0.5% CMC+0.25% Tween 80: 30mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3191 mL | 11.5955 mL | 23.1911 mL | |
| 5 mM | 0.4638 mL | 2.3191 mL | 4.6382 mL | |
| 10 mM | 0.2319 mL | 1.1596 mL | 2.3191 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT04789668
Conditions:Anatomic Stage IV Breast Cancer AJCC v8|Clinical Stage IV Cutaneous Melanoma AJCC v8|Hematopoietic and Lymphoid Cell Neoplasm|Hormone Receptor Positive Breast Adenocarcinoma|Metastatic Lung Non-Small Cell Carcinoma|Metastatic Malignant Neoplasm in the Brain|Metastatic Malignant Solid Neoplasm|Metastatic Melanoma|Metastatic Triple-Negative Breast Carcinoma|Pathologic Stage IV Cutaneous Melanoma AJCC v8|Prognostic Stage IV Breast Cancer AJCC v8|Stage IV Lung Cancer AJCC v8|Stage IVA Lung Cancer AJCC v8|Stage IVB Lung Cancer AJCC v8Link: https://clinicaltrials.gov/ct2/show/NCT01936363
Conditions:Ovarian CancerLink: https://clinicaltrials.gov/ct2/show/NCT00982865
Conditions:Solid Tumors|Cancer
Title:Combination Trial of Pimasertib (MSC1936369B) With Temsirolimus
Status:Terminated
updateDate:2018-07-30
Ctid:NCT01378377
Link: https://clinicaltrials.gov/ct2/show/NCT01378377
Conditions:Advanced Solid TumorLink: https://clinicaltrials.gov/ct2/show/NCT01693068
Conditions:N-Ras Mutated Locally Advanced or Metastasis Malignant Cutaneous MelanomaLink: https://clinicaltrials.gov/ct2/show/NCT01668017
Conditions:Advanced Solid Tumors|Hepatocellular CarcinomaLink: https://clinicaltrials.gov/ct2/show/NCT00957580
Conditions:Leukemia, Myeloid, Acute|Hematologic NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT01713036
Conditions:Locally Advanced or Metastatic Solid TumorsLink: https://clinicaltrials.gov/ct2/show/NCT01992874
Conditions:NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT01016483
Conditions:Pancreatic AdenocarcinomaLink: https://clinicaltrials.gov/ct2/show/NCT01390818
Conditions:Locally Advanced Solid Tumor|Metastatic Solid Tumor|Breast Cancer|Non Small Cell Lung Cancer|Melanoma|Colorectal CancerLink: https://clinicaltrials.gov/ct2/show/NCT01085331
Conditions:Metastatic Colorectal CancerLink: https://clinicaltrials.gov/ct2/show/NCT01357330
Conditions:Solid TumorsLink: https://clinicaltrials.gov/ct2/show/NCT01985191
Conditions:Neoplasm Malignant
|
|
|
|
|
|
|
 MEK1 P124L Recombinant Human Active Protein Kinase
MEK1 P124L Recombinant Human Active Protein Kinase
 MEK1 F53L Recombinant Human Active Protein Kinase
MEK1 F53L Recombinant Human Active Protein Kinase
 MEK5 Recombinant Human Active Protein Kinase
MEK5 Recombinant Human Active Protein Kinase
 MEK1 SESE Recombinant Human Active Protein Kinase
MEK1 SESE Recombinant Human Active Protein Kinase
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA







 463611831
463611831
