| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 5g |
|
||
| 10g |
|
||
| Other Sizes |
|

| 靶点 |
PFV IN with the S217H alteration has an IC50 of 900 nM, making it ten times less sensitive to raltegravir. PFV IN exhibited 10% of WT's activity and was inhibited by Raltegravir at an IC50 of 200 nM, suggesting that PFV IN is less sensitive to IN strand transfer inhibitors (INSTIs) than WT IN is. Similar to the WT enzyme, S217Q PFV IN is also susceptible to raltegravir [1]. Glucuronidation, not the liver, is the mechanism of raltegravir metabolism. With a 95% inhibitory concentration of 31±20 nM in human T cell cultures, raltegravir demonstrates strong anti-HIV-1 action in vitro. Raltegravir exhibited anti-HIV-2 activity in CEMx174 cells as well, with an IC95 of 6 nM. Glucuronidation is the main mechanism of raltegravir metabolism. Strong glucuronidase UGT1A1 inducers should not be utilized since they drastically lower raltegravir concentrations. Hepatic cytochrome P450 activity is only slightly inhibited by raltegravir. Neither CYP3A4-dependent testosterone 6-beta-hydroxylase activity nor CYP3A4 RNA expression are induced by raltegravir [2]. Magnesium and calcium have been shown to decrease raltegravir's cellular permeability [3]. Effectively preventing viral replication is possible with raltegravir and related HIV-1 integrase (IN) strand transfer inhibitors (INSTIs) [4]. Latisavue successfully suppressed SIVmac251 replication in the acutely infected human lymphoid CD4+ T cell lines MT-4 and CEMx174, suggesting an EC90 in the low nanomolar range [5].
|
|---|---|
| 体外研究 (In Vitro) |
具有 S217H 改变的 PFV IN 的 IC50 为 900 nM,使其对拉替拉韦的敏感性降低十倍。 PFV IN 表现出 WT 10% 的活性,并被 Raltegravir 抑制,IC50 为 200 nM,表明 PFV IN 对 IN 链转移抑制剂 (INSTI) 的敏感性低于 WT IN。与 WT 酶类似,S217Q PFV IN 也对拉替拉韦敏感 [1]。拉替拉韦的代谢机制是葡萄糖醛酸化,而不是肝脏。拉替拉韦在人类 T 细胞培养物中的 95% 抑制浓度为 31±20 nM,在体外表现出强大的抗 HIV-1 作用。 Raltegravir 在 CEMx174 细胞中也表现出抗 HIV-2 活性,IC95 为 6 nM。葡萄糖醛酸化是拉替拉韦代谢的主要机制。不应使用强葡萄糖醛酸酶 UGT1A1 诱导剂,因为它们会大大降低拉替拉韦浓度。拉替拉韦仅轻微抑制肝细胞色素 P450 活性。拉替拉韦既不诱导 CYP3A4 依赖性睾酮 6-β-羟化酶活性,也不诱导 CYP3A4 RNA 表达 [2]。镁和钙已被证明可以降低拉替拉韦的细胞通透性[3]。拉替拉韦和相关的 HIV-1 整合酶 (IN) 链转移抑制剂 (INSTI) 可以有效防止病毒复制 [4]。 Latisavue 成功抑制了急性感染的人淋巴 CD4+ T 细胞系 MT-4 和 CEMx174 中的 SIVmac251 复制,表明 EC90 在低纳摩尔范围内 [5]。
在体外实验中,Raltegravir 有效抑制野生型(WT)原型泡沫病毒(PFV)整合酶的链转移活性,其IC50为90 nM。[1] 携带S217H替换(对应于HIV-1的Q148H)的PFV整合酶对Raltegravir的敏感性降低,IC50约为900 nM,表明其效力相比野生型酶降低了10倍。[1] 携带S217Q替换的PFV整合酶对Raltegravir仍然敏感,IC50为40 nM。[1] 携带N224H替换(对应于HIV-1的N155H)的PFV整合酶对Raltegravir的敏感性也降低,IC50为200 nM,表明效力降低了约2倍。[1] |
| 体内研究 (In Vivo) |
随着感染的进展,Ratetelevi 可改善感染 SIVmac251 的非人灵长类动物的病毒免疫状态。拉替拉韦单一疗法导致一种非人类灵长类动物的病毒载量检测不到[5]。
在SIVmac251感染的猕猴(第1组)中,使用raltegravir(50或100 mg,每日两次随食物口服)进行为期十天的单药治疗,导致所有动物的病毒载量显著下降(P = 0.031)和CD4+ T细胞计数显著增加(P = 0.017)。100 mg组中的一只动物达到了检测不到的病毒载量(<40拷贝/mL)。[5] 从第11天开始,在raltegravir治疗方案(所有动物转为100 mg每日两次)中加入两种逆转录酶抑制剂——替诺福韦(PMPA, 20 mg/kg/天,皮下注射)和恩曲他滨(FTC, 50 mg/kg/天,皮下注射)后,病毒载量继续下降,所有动物在两周内达到检测不到的水平,并持续到随访结束(第52天)。CD4计数持续增加,恢复至接种前水平。[5] 在第二组SIVmac251感染的猕猴(第2组)中,raltegravir单药治疗(100 mg每日两次)七天,与治疗前历史值相比,也导致病毒载量显著下降,证实了可重复的抗病毒效果。一只在单药治疗后病毒载量检测不到的动物在治疗暂停后出现反弹。[5] 尽管联合治疗使病毒载量抑制到检测不到的水平,但PBMCs中的原病毒DNA水平保持稳定,治疗52天后没有显著变化,表明慢病毒储存库持续存在。[5] |
| 酶活实验 |
进行了定量的PFV整合酶链转移实验。反应体系(40 µL)包含0.75 µM纯化的PFV整合酶、0.75 µM供体DNA(模拟预处理的病毒U5 DNA末端的双链寡核苷酸)、4 nM(300 ng)超螺旋质粒靶DNA、5 mM MgSO₄(作为二价金属辅因子)、4 µM ZnCl₂、125 mM NaCl、10 mM DTT、25 mM Bis-Tris propane-HCl缓冲液(pH 7.4)和0.8% DMSO。在指定浓度下将Raltegravir加入反应中。通过添加PFV整合酶启动反应,在37°C下孵育1小时,然后加入EDTA和SDS终止反应。对反应产物进行去蛋白化、乙醇沉淀,并在用溴化乙锭染色的1.5%琼脂糖凝胶上进行分离以进行可视化。对于定量,使用特异性引物和荧光DNA结合染料通过定量实时PCR测量整合产物。使用野生型PFV整合酶反应(无抑制剂)的连续稀释液生成的标准曲线来量化抑制剂处理样品中的整合效率。[1]
|
| 细胞实验 |
Caco-2单层渗透性实验: 将Caco-2细胞接种于聚碳酸酯膜Transwell上,培养21天形成单层。通过跨上皮电阻(TEER >600 Ω)和对[14C]甘露醇的低渗透性确认单层完整性。对于渗透性研究,改变顶端隔室的pH(使用如MES用于较低pH,HEPES/Tricine用于较高pH的缓冲液),而底外侧隔室保持pH 7.4。将raltegravir(用于pH效应研究为50 µM;用于金属/奥美拉唑研究为1 µM)加入顶端(用于顶端到底外侧运输)或底外侧(用于底外侧到顶端运输)隔室。平板在37°C、5% CO2下孵育。在不同时间点(例如0、30、60、90、120分钟)从接收隔室取样并用新鲜缓冲液替换。通过LC-MS/MS或闪烁计数分析raltegravir浓度。计算表观渗透性(Papp)和外排比。
细胞积累实验: 将Caco-2细胞接种于6孔板中,培养5天。洗涤细胞并用pH缓冲的孵育溶液(pH 5-8)平衡。加入raltegravir(1 µM),伴或不伴ABCB1抑制剂tariquidar(300 nM)的预孵育和共孵育。在37°C、5% CO2下孵育10分钟后,取细胞外样品,用冰缓冲液洗涤细胞,用水裂解。用乙腈处理细胞裂解物,离心,通过LC-MS/MS分析上清液中的raltegravir含量。[3] |
| 动物实验 |
印度恒河猴经黏膜接种(直肠内或阴道内)300 MID50 致病性 SIVmac251 病毒。感染后第 12 周病毒载量趋于稳定。药物治疗方面,动物被随机分为两组,分别接受口服拉替拉韦单药治疗,剂量为 50 mg 或 100 mg,每日两次,与食物同服,持续 10 天。第 11 天,最初服用 50 mg 的动物改为每日两次服用 100 mg,所有动物除服用拉替拉韦外,还接受皮下注射替诺福韦(PMPA,20 mg/kg/天)和恩曲他滨(FTC,50 mg/kg/天)。治疗持续至第 52 天。定期采集血浆样本,通过定量 RT-PCR 检测病毒载量。收集外周血单核细胞 (PBMC) 用于通过 PCR 定量前病毒 DNA 和进行 CD4+ T 细胞计数流式细胞术分析。同时监测临床化学和血液学指标。[5] |
| 药代性质 (ADME/PK) |
拉替拉韦的溶解度取决于 pH 值。在 10 mM 浓度下,该药物在 pH 6.6 及以下部分不溶,但在 pH 6.8 至 8 时完全溶解。
使用辛醇-水分配法测定的拉替拉韦的亲脂性 (log P) 随着 pH 值从 5 升高到 9 而从约 1.06 降至 -1.29。 使用紫外光谱法测定拉替拉韦的 pKa 值为 6.7。 体外,拉替拉韦在 Caco-2 单层细胞中的细胞渗透性(从顶端到基底外侧)随着细胞外 pH 值的升高而显著降低(从 pH 5 时的 27.3 x 10⁻⁶ cm/s 降至 pH 8.5 时的 2.9 x 10⁻⁶ cm/s)。 二价阳离子(Mg²⁺、Ca²⁺)会降低体外细胞渗透性。 拉替拉韦。 引用的临床研究表明,与奥美拉唑(提高胃pH值)合用可增加健康志愿者体内拉替拉韦的AUC和Cmax,而含镁/铝的抗酸剂可降低拉替拉韦的C12。 拉替拉韦的主要代谢途径是通过UGT1A1进行葡萄糖醛酸化。它是药物转运蛋白ABCB1、SLC22A6和SLC15A1的弱底物。[3] |
| 毒性/毒理 (Toxicokinetics/TK) |
拉替拉韦并非主要细胞色素P450酶的底物或抑制剂。
存在药物相互作用:阿扎那韦(UGT1A1抑制剂)可增加拉替拉韦的暴露量,而利福平(UGT1A1诱导剂)则可降低其暴露量。 与含镁/铝的抗酸剂合用后,75%的受试者拉替拉韦C12低于IC95(50%人血清中为15 ng/mL)。[3] |
| 参考文献 | |
| 其他信息 |
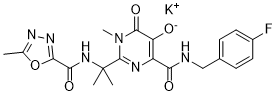
拉替拉韦(商品名:Isentress 和 Isentress HD)是一种处方药,经美国食品药品监督管理局 (FDA) 批准用于治疗成人和儿童的 HIV 感染。拉替拉韦的一种剂型 Isentress 获准用于体重至少 2 公斤(4 磅 4 盎司)的成人和儿童。另一种剂型 Isentress HD 获准用于体重至少 40 公斤(88 磅)的成人和儿童。拉替拉韦必须与其他 HIV 药物联合使用。拉替拉韦钾是一种口服生物利用度高的钾盐,属于人类免疫缺陷病毒 (HIV) 整合酶链转移抑制剂 (HIV-1 INSTI),具有抗 HIV-1 病毒活性。拉替拉韦可与整合酶结合并抑制其活性。整合酶是 HIV 的一种酶,负责将病毒遗传物质插入受感染的人类细胞的遗传物质中。抑制整合酶可阻止 HIV DNA 插入人类 DNA 基因组,从而阻断 HIV 复制。
一种吡咯烷酮衍生物和 HIV 整合酶抑制剂,与其他抗 HIV 药物联合用于治疗 HIV 感染。 另见:拉替拉韦(具有活性部分);拉米夫定;拉替拉韦钾(成分)。 药物适应症 伊生特瑞斯适用于与其他抗逆转录病毒药物联合用于治疗人类免疫缺陷病毒 (HIV-1) 感染。 拉替拉韦是首个获准用于临床治疗 HIV-1 感染的整合酶链转移抑制剂 (INSTI)。[1] 原型泡沫病毒 (PFV) 整合酶体(整合酶-DNA 复合物)与拉替拉韦结合的晶体结构揭示了其结合模式。拉替拉韦(Raltegravir)与整合酶的活性位点结合,通过其药效团中的三个杂原子螯合两个催化二价金属离子(Mg²⁺或Mn²⁺)。这种结合将病毒DNA的3'末端腺苷核苷酸从活性位点置换出来,从而使整合酶失活并阻断链转移反应。[1] 一个关键的相互作用是拉替拉韦的恶二唑环与PFV整合酶中Tyr212(相当于HIV-1整合酶中的Tyr143)的侧链之间形成面对面的π-π堆积。已知该位置的氨基酸替换(例如HIV-1中的Y143H/R/C)会导致耐药性,这是由于这种稳定相互作用的丧失所致。 [1] 该研究以PFV整合酶为模型,旨在了解HIV-1整合酶中的耐药突变。PFV整合酶中的S217H和N224H突变(类似于HIV-1中的Q148H和N155H)会降低其对拉替拉韦的敏感性。突变型整合酶体的晶体结构显示,这些氨基酸替换需要活性位点发生不利的构象重排才能与抑制剂结合,从而解释了其耐药机制。具体而言,S217H(Q148H)突变需要DNA骨架发生显著位移才能与抑制剂结合,而N224H(N155H)突变则破坏了天冬酰胺与DNA骨架之间的稳定相互作用,这种相互作用在抑制剂结合时必须被打破。 [1] PFV整合酶209位丝氨酸的存在(相当于HIV-1的Gly140)可能解释了PFV单突变体S217H为何表现出与HIV-1 Q148H/G140S双突变体相似的高水平耐药性。在S217H突变体的结构中,Ser209与His217形成氢键,这表明在药物选择压力下,HIV-1整合酶140位和148位残基之间可能存在相互作用和协同进化。[1] |
| 分子式 |
C20H20FKN6O5
|
|---|---|
| 分子量 |
482.51
|
| 精确质量 |
482.111
|
| CAS号 |
871038-72-1
|
| 相关CAS号 |
Raltegravir;518048-05-0;Raltegravir-d3 potassium;1246816-98-7;Raltegravir sodium;1292804-07-9
|
| PubChem CID |
23668479
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.46 g/cm3
|
| 熔点 |
282ºC
|
| LogP |
2.131
|
| tPSA |
155.07
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
843
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
IFUKBHBISRAZTF-UHFFFAOYSA-M
|
| InChi Code |
InChI=1S/C20H21FN6O5.K/c1-10-25-26-17(32-10)16(30)24-20(2,3)19-23-13(14(28)18(31)27(19)4)15(29)22-9-11-5-7-12(21)8-6-11;/h5-8,28H,9H2,1-4H3,(H,22,29)(H,24,30);/q;+1/p-1
|
| 化学名 |
potassium;4-[(4-fluorophenyl)methylcarbamoyl]-1-methyl-2-[2-[(5-methyl-1,3,4-oxadiazole-2-carbonyl)amino]propan-2-yl]-6-oxopyrimidin-5-olate
|
| 别名 |
MK-0518; MK0518; MK 0518; MK-0518 potassium; Raltegravir; trade name: Isentress
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.31 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.31 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (4.31 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 30% PEG400+0.5% Tween80+5% Propylene glycol : 30 mg/mL 配方 5 中的溶解度: 25 mg/mL (51.81 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0725 mL | 10.3625 mL | 20.7250 mL | |
| 5 mM | 0.4145 mL | 2.0725 mL | 4.1450 mL | |
| 10 mM | 0.2072 mL | 1.0362 mL | 2.0725 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
 |
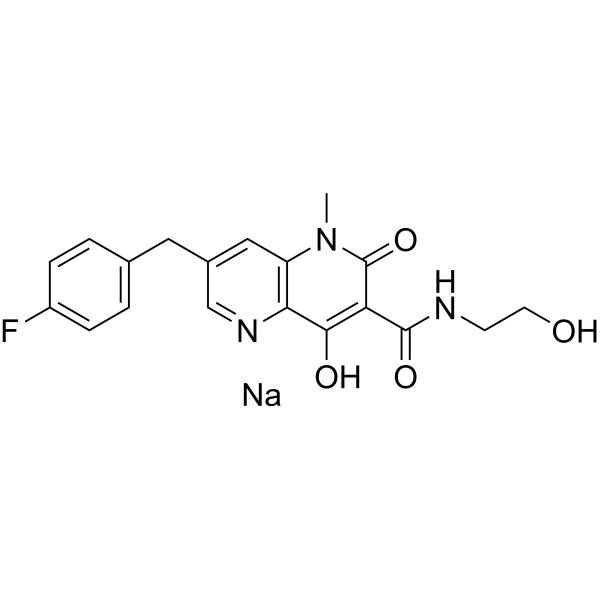
 GSK-364735 potassium
GSK-364735 potassium
 GSK-364735 sodium
GSK-364735 sodium
 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide
1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide
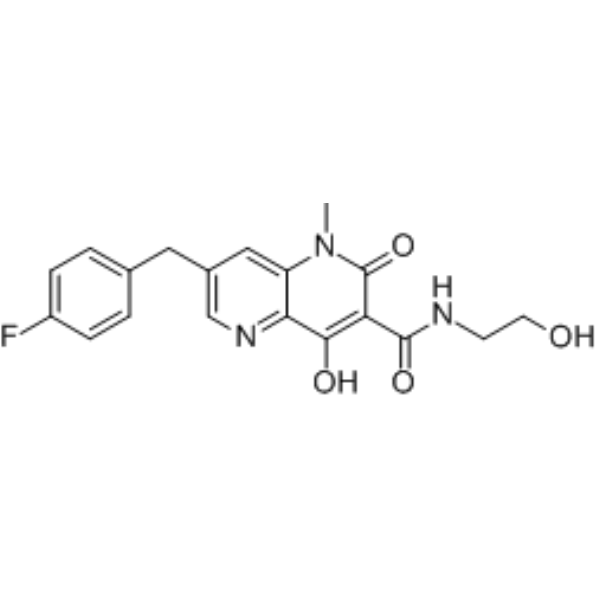 GSK-364735
GSK-364735
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA

 463611831
463611831
