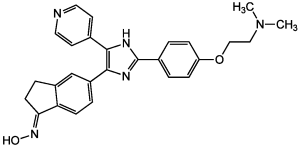
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
B-Raf (Ki = 0.16 nM); c-Raf (Ki = 1.72 nM)
SB590885 is a selective inhibitor of oncogenic mutant B-Raf kinase (BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ). In recombinant human BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ kinase assays, it exhibits an IC₅₀ of 6 nM; it has low activity against wild-type BRAF (BRAFʷᵗ, IC₅₀ = 450 nM) and CRAF (IC₅₀ = 320 nM) [2] - In human BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ-positive A375 melanoma cells, SB590885 inhibits phosphorylated ERK1/2 (p-ERK, downstream of BRAF) with an EC₅₀ of 40 nM [1] - SB590885 shows no significant inhibition of non-RAF kinases, including MEK1 (IC₅₀ > 1 μM), EGFR (IC₅₀ > 5 μM), and PDGFRβ (IC₅₀ > 10 μM) [2] |
|---|---|
| 体外研究 (In Vitro) |
SB590885 对 B-Raf 表现出显着优于 c-Raf 的选择性,Ki 为 0.16 nM 超过 1.72 nM。之前描述的 Raf/VEGFR 激酶抑制剂 BAY 439006(突变体 B-Raf 的 Ki = 38 nM,c-Raf 的 Ki = 6 nM)作为抑制剂不如 SB-590885 有效。 SB590885 对超过 46 种不同的激酶表现出很强的选择性。与 SB590885 一样,多激酶抑制剂 BAY43-9006 不能将致癌 B-Raf 激酶结构域稳定在活性状态。在表达致癌 B-RafV600E 的 Colo205、HT29、A375P、SKMEL28 和 MALME-3M 细胞中,SB590885 处理有效抑制 ERK 磷酸化,EC50 值分别为 28 nM、58 nM、290 nM、58 nM 和 190 nM。它还始终抑制增殖,EC50 值为 0.1 μM、0.87 μM、0.37 μM、0.12 μM 和 0.15 μM。 SB590885 选择性抑制黑色素瘤细胞系的贴壁依赖性生长。 [1] SB590885 的 Kd 为 0.3 nM,对 B-Raf 表现出高亲和力。 [2] 大多数具有 BRAF V600E 突变且无 CDK4 突变的黑色素瘤细胞系(451Lu、WM35 和 WM983)对 SB590885 非常敏感,IC50 <1 μM。在 B-Raf V600E 突变的黑色素瘤中,基因组扩增导致的细胞周期蛋白 D1 水平升高是导致药物耐药性的原因。 [3]
BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ黑色素瘤细胞增殖抑制:在人BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ阳性黑色素瘤细胞系(A375、SK-MEL-28)中,SB590885(0.001-10 μM)浓度依赖性抑制增殖:A375细胞(IC₅₀=30 nM)、SK-MEL-28细胞(IC₅₀=50 nM)。在A375细胞中,0.1 μM剂量使p-ERK水平降低90%(Western blot)、p-MEK水平降低85%,阻断MAPK通路[1] - BRAFʷᵗ细胞不敏感性:在人BRAFʷᵗ黑色素瘤细胞(SK-MEL-5)和正常人包皮成纤维细胞(NHFF)中,SB590885(最高10 μM)抗增殖活性微弱(活力降低<15%),证实其对突变型BRAF的选择性[1] - Cyclin D1介导的耐药性:在过表达cyclin D1的A375细胞(BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ阳性)中,SB590885 抗增殖活性降低:IC₅₀从亲本细胞的30 nM升至180 nM。0.1 μM时克隆形成抑制率仅30%(亲本细胞中为80%),而p-ERK抑制率无显著变化(仍为90%)[3] - 敏感细胞凋亡诱导:在亲本A375细胞中,SB590885(0.1-0.5 μM)浓度依赖性诱导凋亡:0.5 μM使Annexin V⁺细胞比例从溶媒组的4%升至35%,伴随caspase-3和PARP剪切(Western blot)[1] |
| 体内研究 (In Vivo) |
在由表达突变 B-Raf 的 A375P 黑色素瘤细胞创建的小鼠异种移植物中,SB-590885 给药可显着且适度地抑制肿瘤生长[1]。
在三维培养中,长时间暴露于SB-590885可抑制细胞转化和增殖。A, SB-590885在表达B-RafV600E的肿瘤细胞系中选择性抑制锚定非依赖性肿瘤菌落生长(光镜,4倍)。B,通过在网格上一式三份计数单个菌落并对每个细胞系归一化到载体对照(±SE)来量化非锚定生长。C,将HT-1080、WM-NCI和WM-3434的球形培养物用SB-590885处理72小时,并通过光镜(上图)评估侵入性,使用“活/死细胞试验”评估代谢活性,分别用绿色和红色荧光图像表示。D, SB-590885治疗后A375P细胞的致瘤性降低。点,单独用载药剂(▪)或SB-590885 (X)治疗的动物队列的肿瘤生长情况;酒吧、±SE。小鼠治疗21天,之后,用SB-590885治疗的小鼠再观察14天。[1] A375黑色素瘤异种移植模型:在荷A375 BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ异种移植瘤的雌性裸鼠中,口服SB590885(50、100 mg/kg/天,每日一次)剂量依赖性抑制肿瘤生长:100 mg/kg在第21天较溶媒组减少肿瘤体积70%,无显著体重下降(<5%)。肿瘤p-ERK水平降低80%(免疫组化),肿瘤细胞凋亡增加3倍(TUNEL染色)[1] - BRAFʷᵗ肿瘤不敏感性:在荷BRAFʷᵗ SK-MEL-5异种移植瘤的裸鼠中,口服SB590885(100 mg/kg/天)无显著肿瘤抑制作用(体积减少<10%),与其体外选择性一致[1] |
| 酶活实验 |
SB-590885 是一种有效的 B-Raf 抑制剂,Ki 为 0.16 nM,对 B-Raf 的选择性比 c-Raf 高 11 倍,且对其他人类激酶没有抑制作用。
SB-590885(33)是一种有效的、极具选择性的B-Raf激酶抑制剂,是在对一系列咪唑基导联剂的SAR进行评估后确定的。SB-590885是研究B-Raf在神经退行性和其他疾病状态中的作用的理想分子。[2] 化合物活性的确定是通过测试化合物抑制B-Raf介导的激酶死MEK磷酸化的能力(数据以IC50表示)或通过荧光配体置换试验得出的B-Raf结合亲和力(FP,数据以Kd表示)。有关化验细节,请参见Dean, D. K.; Takle, A. K.; Wilson, D. M. PCT Int. Appl. WO 02/24680. 致癌BRAF等位基因是细胞转化的必要和充分条件,这表明化学抑制激活的突变蛋白激酶可能逆转肿瘤表型。在这里,我们报告了SB-590885的特性,这是一种新的三芳基咪唑,它选择性地抑制Raf激酶,对B-Raf的效力比c-Raf的效力更强。晶体学分析显示,SB-590885稳定了致癌的B-Raf激酶结构域的活性结构,这与先前报道的多激酶抑制剂BAY43-9006的作用机制不同。当暴露于SB-590885时,表达致癌B-Raf的恶性细胞表现出对丝裂原激活的蛋白激酶激活、增殖、转化和致瘤性的选择性抑制,而其他癌细胞系和正常细胞对类似治疗表现出不同的敏感性或抗性。这些研究支持了致癌B-Raf作为癌症治疗靶点的有效性,并首次提供了致癌BRAF等位基因表达与选择性B-Raf抑制剂阳性反应之间相关性的证据。[1] 重组BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ激酶实验:将重组人BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ、BRAFʷᵗ或CRAF蛋白(50 ng/孔)与激酶缓冲液(25 mM Tris-HCl pH7.5、10 mM MgCl₂、1 mM DTT、20 μM ATP)、生物素化MEK1衍生肽(底物,2 μM)及不同浓度的SB590885(0.001-100 μM)在30°C孵育60 min。采用均相时间分辨荧光(HTRF)法(铕标记抗磷酸化MEK抗体+链霉亲和素-别藻蓝蛋白)检测磷酸化底物,激酶活性归一化为溶媒对照组,通过非线性回归计算IC₅₀值[2] |
| 细胞实验 |
贴壁细胞增殖分析[3]
将细胞以2.5 × 104个/毫升的密度镀于96孔板中,静置生长过夜。用SB-590885处理细胞三次,72h后,用3-(4,5-二甲基噻唑-2-基)-2,5-二苯四唑溴化试验检测生长抑制水平。数据为至少3次独立实验的平均值±SE。为了确定cyclin D1和CDK4过表达在抗SB-590885中的作用,451Lu细胞分别感染cyclin D1或CDK4病毒。选择过表达cdk4的细胞进行puromycin处理48小时。经Western blotting证实感染后,将细胞进行如上所述的3-(4,5-二甲基噻唑-2-基)-2,5-二苯基溴化四唑测定。[3] 细胞周期分析[3] 细胞以60%的合流度置于10 cm培养皿中,培养过夜,然后用SB-590885(1和3 μmol/L)处理24 h。细胞用乙醇固定,碘化丙啶染色,按上述方法进行分析。在一些实验中,在用SB-590885处理和细胞周期分析之前,细胞感染了CDK4、cyclin D1或CDK4 + cyclin D1的病毒。 [3] 对于增殖测试,细胞用 0.1% DMSO 中的物质处理,并在 37°C、5% CO2 下孵育 72 小时。 Victor 2V 读板器和 CellTiter-Glo 试剂用于测量活细胞数量。根据制造商提供的说明,准备细胞用于 Becton Dickinson FACScan 上的细胞周期分析。使用软件 CellQuest v3.3 收集和检查数据。在琼脂层中使用抑制剂或 DMSO 载体,如前所述进行不依赖贴壁的生长测定。在总共 28 天的时间里,每 5 至 7 天再次向培养物添加培养基、抑制剂或 DMSO。利用标准光学显微镜,观察菌落,用相机捕获,并通过在网格上重复计数三次来定量。 黑色素瘤细胞增殖实验:A375/SK-MEL-28/SK-MEL-5/NHFF细胞以5×10³个细胞/孔接种于96孔板,用含10% FBS的DMEM培养。贴壁24 h后加入SB590885(0.001-10 μM),孵育72 h。MTT法(570 nm吸光度)检测细胞活力,GraphPad Prism计算IC₅₀值[1] - p-ERK/p-MEK Western Blot实验:A375细胞(亲本或过表达cyclin D1)以2×10⁵个细胞/孔接种于6孔板,用SB590885(0.01-0.5 μM)处理24 h。细胞用含蛋白酶/磷酸酶抑制剂的RIPA缓冲液裂解,裂解液(20 μg蛋白/泳道)经SDS-PAGE分离后,膜用抗p-ERK、抗总ERK、抗p-MEK、抗总MEK、抗剪切型caspase-3或抗PARP抗体孵育,随后加入HRP标记二抗,化学发光法显影条带[1,3] - 克隆形成实验:过表达cyclin D1的A375细胞和亲本细胞以1×10³个细胞/孔接种于6孔板,用SB590885(0.01-0.5 μM)处理14天。克隆用甲醇固定、结晶紫染色后计数,相对于溶媒对照组计算克隆形成抑制率[3] - 凋亡实验:亲本A375细胞经SB590885(0.1-0.5 μM)处理48 h后收集,Annexin V-FITC/PI双染色,流式细胞术量化凋亡细胞(Annexin V⁺/PI⁻和Annexin V⁺/PI⁺)比例[1] |
| 动物实验 |
腹腔注射后,评估了SB-590885的药代动力学特征和安全性。结果发现,每日注射50 mg/kg即可达到治疗浓度,且仅引起轻微的体重变化。使用8至12周龄的雌性裸鼠构建肿瘤模型。通过静脉注射5×10⁶个A375P细胞悬浮于Matrigel基质胶中诱导肿瘤。三周后,当肿瘤体积达到150至250 mm³时,将小鼠随机分为8组,并给予相应的治疗。动物分别接受赋形剂(2% N,N-二甲基乙酰胺、2% Cremophor EL和96%酸化水(pH 4-5))或含50 mg/kg SB-590885的赋形剂(每日一次,连续21天)治疗。然后,在治疗结束后14天,再次观察接受SB-590885治疗的小鼠组。每周两次,使用游标卡尺测量肿瘤体积,持续55天。
A375/SK-MEL-5异种移植方案:将A375(5×10⁶个细胞/只)或SK-MEL-5(1×10⁷个细胞/只)溶于Matrigel(1:1 v/v)中,皮下注射到雌性裸鼠(6-7周龄,18-22克)的右侧腹部。当肿瘤体积达到 100–120 mm³ 时,将小鼠随机分为 3 组(每组 n=7):载体组(0.5% 甲基纤维素 + 0.1% Tween 80,口服)、SB590885 50 mg/kg 组(口服,每日一次)、SB590885 100 mg/kg 组(口服,每日一次)。药物每日给药,持续 21 天。每 3 天测量一次肿瘤体积(V = π×L×W²/6)和体重。研究结束时,切除肿瘤:一半用福尔马林固定,用于 p-ERK 免疫组织化学和 TUNEL 染色;另一半冷冻保存,用于蛋白质提取 [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
血浆蛋白结合率:在小鼠血浆中(通过超滤法测定),SB590885 在 0.01–1 μM 浓度范围内具有约 90% 的蛋白结合率,且与浓度无关 [1]
- 急性毒性:在接受 SB590885 治疗的裸鼠中(剂量高达 100 mg/kg/天,持续 21 天),未观察到死亡或严重毒性。体重保持稳定,血清 ALT/AST(肝脏标志物)和肌酐(肾脏标志物)均在正常范围内 [1] - 正常组织安全性:在 NHFF 细胞(正常成纤维细胞)中,SB590885(浓度高达 10 μM)未显示出明显的细胞毒性(细胞活力降低 <10%),表明其具有良好的治疗指数 [1] |
| 参考文献 |
|
| 其他信息 |
(E)-SB-590885 是一种 N-{5-[2-{4-[2-(二甲氨基)乙氧基]苯基}-4-(吡啶-4-基)-1H-咪唑-5-基]-2,3-二氢-1H-茚-1-亚基}羟胺,其中肟基具有 E 构型。
近期研究表明,黑色素瘤细胞系对 MEK 和 BRAF 抑制剂的反应存在显著的异质性。本研究旨在探讨细胞周期蛋白依赖性激酶 4 (CDK4) 和/或细胞周期蛋白 D1 的失调是否会导致黑色素瘤细胞对 BRAF 抑制剂产生耐药性。突变筛查鉴定出一组同时携带BRAF V600E突变和CDK4突变的黑色素瘤细胞系:K22Q (1205Lu)、R24C (WM39、WM46和SK-Mel-28)以及R24L (WM902B)。药理学研究表明,CDK4突变的存在并不影响这些细胞系对BRAF抑制剂的敏感性。唯一表现出显著BRAF抑制剂耐药性的细胞系同时携带CDK4突变和CCND1扩增。阵列比较基因组杂交分析显示,在17%的BRAF V600E突变的人类转移性黑色素瘤样本中存在CCND1扩增,表明该发现具有临床意义。由于细胞系中CCND1扩增的水平低于临床标本中的水平,我们在一种药物敏感的黑色素瘤细胞系中单独过表达细胞周期蛋白D1,以及在CDK4存在的情况下过表达细胞周期蛋白D1。细胞周期蛋白D1的单独过表达即可增加耐药性,而当细胞周期蛋白D1和CDK4同时过表达时,这种耐药性会进一步增强。总之,基因组扩增导致的细胞周期蛋白D1水平升高可能导致BRAF V600E突变型黑色素瘤对BRAF抑制剂产生耐药性,尤其是在CDK4突变/过表达的情况下。[3] SB590885是一种研究级选择性BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ抑制剂,其开发目的是为了验证靶向突变型BRAF治疗BRAF V600E突变型癌症的潜力。它尚未获准用于临床[1,2] - 作用机制:其抗肿瘤作用是通过特异性抑制BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ激酶活性介导的,从而阻断下游MAPK(RAF-MEK-ERK)信号通路——该通路在BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ阳性癌症中持续激活,驱动细胞增殖和存活[1,2] - 耐药机制:临床前研究表明,BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ黑色素瘤细胞对SB590885的耐药性可由细胞周期蛋白D1过表达介导,细胞周期蛋白D1过表达可绕过MAPK通路抑制以维持细胞周期进程。这凸显了联合治疗策略(例如,BRAF + 细胞周期蛋白 D1 抑制剂)的必要性[3] - 研究意义:SB590885 是早期选择性 BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ 抑制剂之一,为后续 BRAF 抑制剂(例如,维莫非尼)的临床开发提供了基础证据[1,2] |
| 分子式 |
C27H27N5O2
|
|
|---|---|---|
| 分子量 |
453.54
|
|
| 精确质量 |
453.216
|
|
| 元素分析 |
C, 71.50; H, 6.00; N, 15.44; O, 7.06
|
|
| CAS号 |
405554-55-4
|
|
| 相关CAS号 |
|
|
| PubChem CID |
135421339
|
|
| 外观&性状 |
White to yellow solid
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
691.1±55.0 °C at 760 mmHg
|
|
| 闪点 |
371.8±31.5 °C
|
|
| 蒸汽压 |
0.0±2.3 mmHg at 25°C
|
|
| 折射率 |
1.666
|
|
| LogP |
5.01
|
|
| tPSA |
86.63
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
34
|
|
| 分子复杂度/Complexity |
674
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
O([H])/N=C1/C2C([H])=C([H])C(C3=C(C4C([H])=C([H])N=C([H])C=4[H])N([H])C(C4C([H])=C([H])C(=C([H])C=4[H])OC([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])[H])=N3)=C([H])C=2C([H])([H])C/1([H])[H]
|
|
| InChi Key |
MLSAQOINCGAULQ-QFMPWRQOSA-N
|
|
| InChi Code |
InChI=1S/C27H27N5O2/c1-32(2)15-16-34-22-7-3-19(4-8-22)27-29-25(18-11-13-28-14-12-18)26(30-27)21-5-9-23-20(17-21)6-10-24(23)31-33/h3-5,7-9,11-14,17,33H,6,10,15-16H2,1-2H3,(H,29,30)/b31-24+
|
|
| 化学名 |
(NE)-N-[5-[2-[4-[2-(dimethylamino)ethoxy]phenyl]-5-pyridin-4-yl-1H-imidazol-4-yl]-2,3-dihydroinden-1-ylidene]hydroxylamine
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.51 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.51 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.51 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 2% Cremophor EL, 2% N,N-dimethylacetamide, pH 5.0: 30mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2049 mL | 11.0244 mL | 22.0488 mL | |
| 5 mM | 0.4410 mL | 2.2049 mL | 4.4098 mL | |
| 10 mM | 0.2205 mL | 1.1024 mL | 2.2049 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Overexpression of cyclin D1 reduces sensitivity to the BRAF inhibitor SB590885.Mol Cancer Ther.2008 Sep;7(9):2876-83. |
Identification of human melanoma samples and a cell line with high levels of cyclin D1 (CCND1) amplification.A, DNA copy number profiles of chromosome 11 for melanoma tumor samples TB2668F1, TB2673F1, and the melanoma cell line WM39.Mol Cancer Ther.2008 Sep;7(9):2876-83. |
Melanoma cell lines withCDK4mutations are not resistant to SB590885.A, melanoma cell lines with aCDK4mutation (WM39, WM46, SK-Mel-28, WM902B, WM793, and1205Lu: red, open symbols) and melanoma lines withoutCDK4mutations (WM983, WM164, and451Lu; blue, closed symbols) were treated with increasing concentrations of SB590885 (1 nmol/L – 10 μmol/L) for 72 h before being treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.Mol Cancer Ther.2008 Sep;7(9):2876-83. |
 RAF-IN-2
RAF-IN-2
 BRAF ligand-1
BRAF ligand-1
 CST905
CST905
 BRAFV600E-IN-2
BRAFV600E-IN-2
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
