| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
PI3Kδ (IC50 = 12 nM); GSK-3α (IC50 = 1.5 nM)
A potent, ATP-competitive, and highly selective PI3K inhibitor called seletalisib can prevent AKT phosphorylation after the BCR is activated in a B-cell line. Seletalisib inhibits superoxide release from human neutrophils when N-formyl peptides (fMLP) are stimulated, but not when phorbol myristate acetate (PMA), suggesting that it has a PI3Kδ-specific activity. Neither PBMCs nor other cell types treated with seletalisib show any signs of cytotoxicity. Seletalisib prevents human T cells from producing a number of cytokines after they become activated. The differentiation of T cells into Th1, Th2, and Th17 subtypes is inhibited by seletalisib. Seletalisib also prevents B-cell growth and cytokine production. Seletalisib blocks basophil degranulation induced by anti-IgE and B-cell activation in human whole blood assays[1]. |
|---|---|
| 体外研究 (In Vitro) |
一种名为 seletalisib 的有效、ATP 竞争性、高选择性 PI3K 抑制剂可以在 B 细胞系中 BCR 激活后阻止 AKT 磷酸化。当 N-甲酰肽 (fMLP) 受到刺激时,Seletalisib 会抑制人中性粒细胞释放超氧化物,但当佛波醇肉豆蔻酸酯乙酸酯 (PMA) 受到刺激时,Seletalisib 则不会,这表明它具有 PI3Kδ 特异性活性。用 seletalisib 处理的 PBMC 和其他细胞类型均未显示任何细胞毒性迹象。 Seletalisib 可防止人类 T 细胞在激活后产生多种细胞因子。 seletalisib 抑制 T 细胞分化为 Th1、Th2 和 Th17 亚型。 Seletalisib 还可以阻止 B 细胞生长和细胞因子产生。在人全血检测中,Seletalisib 可阻断抗 IgE 诱导的嗜碱性粒细胞脱粒和 B 细胞激活[1]。
在Ramos人B细胞系中,seletalisib有效抑制了抗IgM诱导的AKT磷酸化,IC50为15 nM(几何平均数;95% CI:9.3–23.5)。[1] Seletalisib抑制fMLP刺激的人中性粒细胞超氧化物释放,IC50为16 nM(几何平均数;95% CI:11–24),但不抑制PMA刺激的超氧化物释放,表明其具有PI3Kδ特异性活性。[1] 在抗CD3刺激的人PBMCs中,seletalisib抑制IFNγ(IC50 = 54 nM)、IL-17(IC50 = 21 nM)和TNFα(IC50 = 31 nM)的分泌。在大鼠PBMCs中,它抑制抗CD3诱导的TNFα释放,IC50为22 nM。[1] Seletalisib抑制屋尘螨(HDM)诱导的过敏供者PBMCs分泌IL-5和IL-13,平均IC50值分别为15 nM和2 nM。[1] Seletalisib抑制抗IgM诱导的人B细胞增殖,IC50为16 nM(范围:10–19 nM)。[1] Seletalisib抑制CpG ODN 2006诱导的纯化人B细胞释放IL-6和IL-10,IC50值分别为57 nM和18.1 nM。[1] 在人全血中,seletalisib抑制抗IgM诱导的B细胞CD69表达,IC50为57 nM(几何平均数;95% CI:22–143)。[1] 在人全血中,seletalisib抑制抗IgE诱导的嗜碱性粒细胞脱颗粒(通过CD63表达测量),IC50为35 nM(几何平均数;95% CI:27–45)。[1] BioMAP分析显示,seletalisib选择性抑制免疫细胞反应(例如,BT共培养系统中的B细胞增殖、细胞因子和IgG产生),在PBMC测定中未显示细胞毒性,在非白细胞系统中也无活性,这与泛PI3K抑制剂形成对比。[1] |
| 体内研究 (In Vivo) |
Seletalisib 显着减少 TCR 刺激后大鼠释放的 IL-2 量。在所有测试剂量下均观察到塞拉利西布的抑制作用,在剂量水平≥1 mg/kg 时几乎达到完全抑制。 seletalisib 的体内作用很强,估计 IC50 值<10 nM[1]。
在Lewis大鼠抗CD3抗体诱导的IL-2释放模型中,在抗CD3攻击前30分钟口服给予seletalisib(0.1–10 mg/kg)可显著且剂量依赖性地抑制IL-2释放。在剂量≥1 mg/kg时几乎达到完全抑制。基于血药浓度-反应关系,估计的体内IL-2抑制IC50 < 10 nM。[1] |
| 酶活实验 |
将 Seletalisib 溶解在 DMSO 中,并在浓度响应 (seletalisib) 中进行测试,以探索 PI3Kδ 特异性抑制与 I 类 PI3K 信号传导完全抑制相比的效果。此外,还在 BioMap BT 细胞系统中检查了 1000、100、10 和 1 nM 的 seletalisib 的作用。根据化合物如何影响细胞因子、生长因子、粘附分子和增殖终点等细胞读数水平,创建活性概况[1]。
使用竞争性时间分辨荧光共振能量转移(TR-FRET)测定法评估seletalisib对PI3K亚型的抑制活性。在该测定中,PI3K酶活性将磷脂酰肌醇二磷酸(PIP2)转化为磷脂酰肌醇三磷酸(PIP3)。生成的PIP3与生物素化的PIP3示踪剂竞争结合GST标记的pleckstrin同源结构域蛋白。该复合物使用链霉亲和素-别藻蓝蛋白和铕标记的抗GST抗体进行检测,接近时产生TR-FRET信号。抑制PI3K会减少PIP3的产生,从而增加TR-FRET信号。实验步骤包括将化合物与PIP2和ATP预孵育,然后加入PI3K酶。孵育后,终止反应,加入检测试剂,测量TR-FRET信号。通过在不同ATP浓度(2、40、200和1000 µM)下进行测定,证实了seletalisib的ATP竞争性。[1] 使用Z’-LYTE FRET和Adapta通用激酶测定法,在10 µM的筛选浓度下进行了激酶选择性分析。[1] |
| 细胞实验 |
将在无血清 RPMI 1640 中连续稀释的塞莱达西布和山羊抗人 F(ab)2 IgM 添加到铺在无血清 RPMI 1640 中的 Ramos 细胞中。将板孵育 10 分钟,置于冰上,然后将细胞离心将它们颗粒化。 MSD 测定用于鉴定细胞磷酸化的 AKT。
Ramos pAKT测定: 将Ramos细胞接种在无血清培养基中,用系列稀释的seletalisib处理,然后用抗IgM抗体刺激。短暂孵育后,裂解细胞,使用微尺度发现(MSD)免疫测定法定量磷酸化AKT(pAKT)水平。[1] 人中粒细胞超氧化物释放测定: 从人血中富集的中性粒细胞在含有超氧化物检测混合物(二氢细胞松弛素B、辣根过氧化物酶、叠氮化钠、Amplex Red)的PBS中与seletalisib一起孵育,并用fMLP或PMA刺激。孵育后,测量荧光强度以定量超氧化物释放。[1] T细胞激活测定(人/大鼠): 从人或大鼠血液中分离PBMCs。对于人测定,用抗CD3抗体包被板。将PBMCs和seletalisib加入板中,孵育48小时。然后收集培养上清液,使用MSD或ELISA试剂盒进行细胞因子分析(人:IFNγ、IL-17、TNFα;大鼠:TNFα)。[1] 屋尘螨(HDM)测定: 将HDM过敏供者的PBMCs与seletalisib和HDM提取物一起孵育6天。收集上清液,通过ELISA测量IL-5和IL-13水平。[1] B细胞增殖测定: 通过CD19阳性选择从PBMCs中分离B细胞,用CFSE染色,并与丝裂霉素C处理的抗原呈递细胞共培养。用seletalisib处理细胞,并用抗IgM、IL-2和IL-10刺激。6天后,通过流式细胞术分析CD19+细胞中CFSE的稀释度以测量增殖。[1] CpG诱导的B细胞因子测定: 纯化的CD19+ B细胞用seletalisib处理,并用CpG ODN 2006刺激48小时。收集上清液,通过ELISA测量IL-6和IL-10水平。[1] 全血CD69 B细胞激活测定: 将人全血等分试样与抗IgM和seletalisib一起孵育20小时。然后用抗CD19和抗CD69抗体染色细胞,裂解,并通过流式细胞术分析CD19+ B细胞上CD69的平均荧光强度。[1] 全血嗜碱性粒细胞脱颗粒测定: 将人全血与seletalisib一起孵育,然后用抗IgE刺激。孵育后,用抗体混合物(CD123、HLA-DR、CD63)染色细胞,裂解,并通过流式细胞术分析。将嗜碱性粒细胞圈定为SSC^lo、CD123+、HLA-DR-细胞,并测量CD63表面表达作为脱颗粒的标志。[1] |
| 动物实验 |
大鼠:大鼠经口灌胃给予 seletalisib(0.1-10 mg/kg,500 μL)或溶剂,30 分钟后静脉注射 200 μL 抗 CD3 抗体。口服和静脉注射的溶剂分别为甲基纤维素和生理盐水。测定 IL-2 和 seletalisib 的水平[1]。
抗 CD3 诱导的 Lewis 大鼠 IL-2 释放:成年雄性 Lewis 大鼠(6-8 周龄)经口灌胃给予 seletalisib(0.1、1、3 或 10 mg/kg)或溶剂(0.5% 甲基纤维素),体积为 500 μL。30 分钟后,大鼠接受静脉注射抗 CD3 抗体(100 μg/kg,200 μL 生理盐水)。阴性对照组同时接受口服和静脉注射溶剂。阳性对照组接受口服赋形剂和静脉注射抗CD3抗体。注射抗CD3抗体90分钟后,对大鼠进行麻醉,通过心脏末端穿刺采集血液至EDTA或肝素抗凝管中,并制备血浆用于ELISA法测定IL-2。同时,血液样本也用于LC-MS/MS法分析seletalisib浓度。[1] |
| 参考文献 | |
| 其他信息 |
Seletalisib 已用于研究原发性干燥综合征的治疗和基础科学的临床试验。
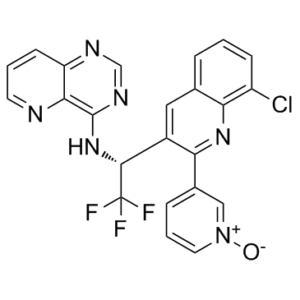
Seletalisib(UCB-...)是一种新型、高效、选择性且与 ATP 竞争的小分子 PI3Kδ 亚型抑制剂。[1] 它选择性抑制 PI3Kδ,而 PI3Kδ 主要表达于白细胞,并位于 B 细胞受体 (BCR)、T 细胞受体 (TCR) 和 Fcε 受体 I (FcεRI) 等免疫受体的下游,这表明其具有治疗 B 细胞恶性肿瘤和自身免疫/炎症性疾病(例如类风湿性关节炎、系统性红斑狼疮、银屑病)的潜力。[1] 它已进入临床试验阶段。截至本文发表时,该药物正在健康志愿者和银屑病患者(NCT02303509)以及原发性干燥综合征患者(NCT02610543)中进行评估。[1] |
| 分子式 |
C23H14CLF3N6O
|
|
|---|---|---|
| 分子量 |
482.85
|
|
| 精确质量 |
482.086
|
|
| 元素分析 |
C, 57.21; H, 2.92; Cl, 7.34; F, 11.80; N, 17.41; O, 3.31
|
|
| CAS号 |
1362850-20-1
|
|
| 相关CAS号 |
|
|
| PubChem CID |
56928390
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.5±0.1 g/cm3
|
|
| 沸点 |
710.8±60.0 °C at 760 mmHg
|
|
| 闪点 |
383.7±32.9 °C
|
|
| 蒸汽压 |
0.0±2.3 mmHg at 25°C
|
|
| 折射率 |
1.692
|
|
| LogP |
2.21
|
|
| tPSA |
89
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
9
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
34
|
|
| 分子复杂度/Complexity |
690
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
ClC1=CC=CC2=C1N=C(C1=CC=C[N+](=C1)[O-])C(=C2)[C@H](C(F)(F)F)NC1C2C(=CC=CN=2)N=CN=1
|
|
| InChi Key |
LNLJHGXOFYUARS-OAQYLSRUSA-N
|
|
| InChi Code |
InChI=1S/C23H14ClF3N6O/c24-16-6-1-4-13-10-15(18(31-19(13)16)14-5-3-9-33(34)11-14)21(23(25,26)27)32-22-20-17(29-12-30-22)7-2-8-28-20/h1-12,21H,(H,29,30,32)/t21-/m1/s1
|
|
| 化学名 |
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.18 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0710 mL | 10.3552 mL | 20.7104 mL | |
| 5 mM | 0.4142 mL | 2.0710 mL | 4.1421 mL | |
| 10 mM | 0.2071 mL | 1.0355 mL | 2.0710 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Seletalisib is a selective, ATP-competitive PI3Kδinhibitor.
Seletalisib is a potent inhibitor of IL-2 release in a rat model of inflammation (primary pharmacology model).J Pharmacol Exp Ther.2017 Jun;361(3):429-440 |
|---|
Seletalisib shows potent and selective cellular inhibition of PI3Kδsignaling.
Seletalisib shows potent activity in human whole blood assays.J Pharmacol Exp Ther.2017 Jun;361(3):429-440 |
Seletalisib shows anti-inflammatory activity in cellular assays of adaptive immunity.J Pharmacol Exp Ther.2017 Jun;361(3):429-440 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA





 463611831
463611831
