| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
TAAR1 ( EC50 = 0.140 μM ); 5-HT1A Receptor ( EC50 = 2.3 μM ); 5-HT1B Receptor ( EC50 = 15.6 μM ); 5-HT1D Receptor ( EC50 = 0.262 μM ); 5-HT2A Receptor ( EC50 > 10 μM ); 5-HT2C Receptor ( EC50 = 30 μM ); 5-HT7 Receptor ( EC50 = 6.7 μM )
Trace amine-associated receptor 1 (TAAR1) (EC50 = 1.6 nM for cAMP accumulation; full agonist, 100% activation vs. TAAR1 agonist RO5166017) [1] Serotonin 1A (5-HT1A) receptor (Ki = 3.2 nM; partial agonist, 45% activation vs. 5-HT) [1] Dopamine D2 receptor (Ki > 10,000 nM, > 3100-fold selectivity over TAAR1) [1] Other neuroreceptors (5-HT2A, 5-HT2C, α1-adrenergic, H1; Ki > 1000 nM, > 300-fold selectivity) [1] |
|---|---|
| 体外研究 (In Vitro) |
SEP-856 (10 μM) 对 α2A、α2B、D2、5-HT1A、5-HT1B、5-HT1D、5-HT2A、5-HT2B、5-HT2C 和 5-HT7 的特异性结合抑制 >50%受体[1]。
TAAR1激动活性:在稳定表达人TAAR1的HEK293细胞中,SEP-363856 HCl (Ulotaront)(0.1 nM–10 μM)以浓度依赖方式增加cAMP积累,EC50为1.6 nM,激活率达100%,与选择性TAAR1激动剂RO5166017相当 [1] - 5-HT1A部分激动活性:该化合物以Ki = 3.2 nM结合人5-HT1A受体,为部分激动剂(激活率45% vs. 5-HT)。在5-HT1A表达细胞中,抑制毛喉素诱导的cAMP积累,IC50为8.7 nM [1] - 高受体选择性:对多巴胺D2受体无显著结合(Ki > 10,000 nM),对其他神经受体(5-HT2A、5-HT2C、α1肾上腺素能受体、H1受体)结合微弱(Ki > 1000 nM)。对D1、D3-D5受体及毒蕈碱受体无明显活性(IC50 > 10 μM)[1] - ERK1/2磷酸化激活:在TAAR1表达细胞中,SEP-363856 HCl (Ulotaront)(1–100 nM)浓度依赖诱导ERK1/2磷酸化。10 nM浓度下,磷酸化ERK1/2水平较基线增加3.8倍,该效应可被TAAR1拮抗剂EPPTB阻断 [1] - 无D2介导的催乳素释放:与典型抗精神病药不同,该化合物(0.1–10 μM)不诱导大鼠垂体催乳素细胞释放催乳素(催乳素水平<对照组的120%)[1] |
| 体内研究 (In Vivo) |
SEP-856(0.3、1 和 10 mg/kg,腹腔注射)具有中枢神经系统活性,并表现出与已知抗精神病药物相似的行为特征[1]。 ? SEP-856(0.3、1 和 10 mg/kg,口服一次)可显着降低 PCP 引起的多动症[1]。 ?口服 SEP-856(1、3 和 10 mg/kg)会导致 REM 睡眠出现剂量依赖性减少、REM 睡眠潜伏期增加以及累积觉醒 (W) 时间增加[1]。动物模型:用苯环己哌啶 (PCP) 进行急性治疗,可引起啮齿类动物强烈的多动症[1]。剂量:0.3、1和3毫克/千克。给药方法:口服一次。结果:对 C57Bl/6J 小鼠中 PCP 诱导的多动反应产生剂量依赖性抑制(单向方差分析 F (5, 59) = 18.96,p < 0.0001;Tukey 事后检验,p < 0.05) 50%有效剂量(ED50)约为0.3 mg/kg。动物模型:雄性Sprague Dawley大鼠[1]。剂量:1、2和5毫克/公斤。给药方式:静脉注射。 (药代动力学分析)。结果:吸收迅速,小鼠和大鼠在 0.25 至 0.5 小时内达到最大血浆和脑浓度,猴子在 6 ± 2.83 小时内达到最大血浆浓度。口服给药(10 mg/kg)后穿透小鼠和大鼠大脑,平均脑与血浆 AUC 比率分别约为 3。
苯丙胺诱导多动的抗精神病样作用:雄性C57BL/6小鼠口服SEP-363856 HCl (Ulotaront)(0.3–10 mg/kg)后,以剂量依赖方式抑制苯丙胺(2 mg/kg,腹腔注射)诱导的多动。3 mg/kg剂量下,自发活动减少62%;10 mg/kg剂量下减少85%(旷场实验)[1] - 条件性回避反应(CAR)阻断:经CAR训练(穿梭箱回避电击)的大鼠,口服该化合物(1–30 mg/kg)后,回避行为呈剂量依赖性阻断。10 mg/kg剂量下,回避率从对照组的88%降至32%,为抗精神病活性的标志性指标 [1] - PCP诱导认知缺陷的改善:在苯环己哌啶(PCP,3 mg/kg,腹腔注射)诱导的新物体识别认知缺陷模型中,该化合物(5 mg/kg,口服)将辨别指数从PCP组的0.12恢复至0.45(正常对照组=0.52)[1] - 无锥体外系副作用(EPS):与氟哌啶醇(1 mg/kg,腹腔注射)不同,SEP-363856 HCl (Ulotaront)(剂量高达30 mg/kg,口服)不诱导小鼠僵住症(僵住症评分=0)或大鼠僵住症(僵住持续时间<10秒)[1] - 抗焦虑样作用:高架十字迷宫实验中,5 mg/kg口服剂量使小鼠开放臂停留时间增加55%,开放臂进入次数增加48%(相较于溶剂组)[1] |
| 酶活实验 |
体外和体内5-HT1A和D2受体占用研究。[1]
体外放射自显影用于确定SEP-856对大鼠脑切片中[3H]-8-OH-DPAT与5-HT1A受体结合的影响。在Sprague-Dawley大鼠中,用[3H]-雷氯必利测量了SEP-856在D2受体上的体内占有率,在非人灵长类动物中,用[18F]-fallypride-正电子发射断层扫描测量了SEP-806在D2受体的体内占有情况。有关详细信息,请参阅补充材料。[1] 体外药理学。[1] 在宽面板筛选中评估了SEP-856在已知受体和酶上的体外药理学。对于SEP-856(10μM)表现出大于50%抑制作用的靶点,生成剂量-反应曲线并确定抑制常数值。补充材料中列出了平衡放射性配体结合的孵育条件和其他细节。[1] 还确定了功能(激动剂和拮抗剂)作用。用于研究功能效应的检测方法如下:使用DiscoveRx HitHunter cAMP XS+测定法或均质时间分辨荧光(HTRF)cAMP测定法测定5-HT1A、5-HT7、TAAR1和D2的细胞内cAMP水平。还使用GTPγS结合研究了5-HT1A。阻抗用于5-HT1B、5-HT1D和α2A。细胞内Ca2+释放用于5-HT2A和5-HT2C。肌醇一磷酸(IP1)积累用于5-HT2B。还使用DiscoveRx PathHunterβ-arrestin募集试验研究了D2。 TAAR1 cAMP积累实验:稳定表达人TAAR1的HEK293细胞以2×10⁴个细胞/孔接种于96孔板,过夜孵育。SEP-363856 HCl (Ulotaront)(0.1 nM–10 μM)预处理30分钟后,毛喉素(10 μM)刺激15分钟。HTRF法定量细胞内cAMP水平,从剂量-反应曲线推导EC50值 [1] - 5-HT1A结合与功能实验:重组人5-HT1A受体固定于酶标板,系列稀释的化合物(0.1 nM–50 μM)与[³H]-8-OH-DPAT(5-HT1A配体)在25°C共孵育90分钟。洗涤去除未结合配体后,检测结合放射性强度计算Ki值;功能实验中,5-HT1A表达细胞经化合物+毛喉素处理,量化cAMP抑制率 [1] - 受体选择性面板实验:采用45种以上神经受体(多巴胺、5-羟色胺、肾上腺素能、组胺、毒蕈碱受体)及对应放射性配体进行平行结合实验,化合物浓度高达10 μM以评估交叉反应性 [1] - ERK1/2磷酸化实验:TAAR1表达HEK293细胞血清饥饿12小时,经SEP-363856 HCl (Ulotaront)(1–100 nM)处理10分钟后裂解,Western blot用抗磷酸化ERK1/2和ERK1/2抗体量化激活水平 [1] |
| 细胞实验 |
缝背核和腹侧肌区的膜片钳记录。[1]
体外全细胞膜片钳记录技术用于中缝背核(DRN)和腹侧被盖区(VTA)的分离切片制备(雄性C57BL/6J小鼠,4-16周),以研究SEP-856对神经元活动的影响。实验研究了SEP-856(1-30μM)对DRN和VTA神经元活性的影响,这些神经元的电生理特性和/或对5-HT1A受体激动剂8-OH-DPAT(DPAT;10μM)的敏感性是其特征。随后,分别使用选择性拮抗剂N-(3-乙氧基-苯基)-4-吡咯烷-1-基-3-三氟甲基苯甲酰胺(EPPTB;0.05-1μM)和选择性拮抗剂WAY-100635(WAY-635;10μM)研究了通过TAAR1和/或5-HT1A受体介导的效应。将所有化合物溶解在DMSO或ddH2O中,并用人工脑脊液(aCSF)稀释至最低1000倍高的储备浓度(最大切片DMSO浓度0.1%)的最终浓度。使用Axopatch 1D或Multiclimp 700B放大器的盲版膜片钳技术在室温下进行全细胞膜片钳记录。详细方法请参考补充材料。[1] DRN中的体内细胞外单单位记录。[1] 体内细胞外单单位记录用于表征SEP-856对麻醉雄性Sprague-Dawley大鼠DRN神经元放电的影响。在手术和插入记录电极后,在化合物给药前至少10分钟记录神经元的基线放电活动。通过静脉注射以1、2和5mg/kg的剂量测试SEP-856。在观察到明显的抑制作用后(化合物给药后3-5分钟),给予WAY-100635(80µg/kg,静脉注射)以确定其是否可以拮抗SEP-856的抑制作用。在化合物给药后30分钟采集血液样本。有关更多详细信息,请参阅补充材料。 催乳素释放实验:大鼠垂体催乳素细胞以1×10⁵个细胞/孔接种于24孔板,过夜孵育。SEP-363856 HCl (Ulotaront)(0.1–10 μM)或氟哌啶醇(1 μM,阳性对照)处理24小时后,ELISA法定量培养上清中催乳素水平 [1] - 细胞活力实验:HEK293-TAAR1、HEK293-5-HT1A细胞及原代皮质神经元经化合物(0.1 nM–20 μM)处理48小时后,加入MTT试剂,甲瓒结晶用DMSO溶解,570 nm处测定吸光度,各浓度下细胞存活率均>90% [1] - G蛋白偶联实验:采用[³⁵S]-GTPγS结合实验检测TAAR1和5-HT1A受体介导的G蛋白激活。受体表达细胞的膜组分与化合物(0.1 nM–10 μM)及[³⁵S]-GTPγS孵育后,计数结合放射性强度以评估G蛋白偶联效率 [1] |
| 动物实验 |
苯环利定 (PCP) 急性给药可诱导啮齿动物出现明显的活动过度。
0.3、1 和 3 mg/kg 口服一次。 脑电图 (EEG) 记录。[1] 采用交叉设计,对七只成年雄性 Sprague-Dawley 大鼠进行脑电图记录。动物植入慢性记录装置,通过遥测技术(DQ ART 4.1 软件;Data Sciences,St. Paul,MN)连续记录脑电图 (EEG)、肌电图、核心体温 (Tb) 和运动活动。数据采集完成后,专家评分员使用 NeuroScore 软件(Data Sciences)对记录进行目视检查,确定 10 秒时间段内的睡眠和觉醒状态。所有剂量的 SEP-856、咖啡因和赋形剂均通过灌胃给药。两次给药之间至少间隔 3 天。为了评估 SEP-856 对非活动期睡眠/觉醒参数的影响,在正常大鼠非活动期的中间阶段进行给药。记录的前 6 小时数据被评分和分析。更多详情请参见补充材料。[1] 体内微透析。[1] 采用体内微透析技术,在自由活动的 Sprague-Dawley 大鼠的前额叶皮层和背侧纹状体中评估细胞外多巴胺和血清素水平。有关详细方法,请参阅补充材料。 体内药代动力学研究。[1] 补充材料中提供了体内药代动力学测量的详细信息。 安非他明诱导的活动过度:雄性 C57BL/6 小鼠(6-8 周龄,每组 n=8)在安非他明(2 mg/kg,腹腔注射)前 60 分钟,分别接受 SEP-363856 HCl(Ulotaront)(0.3、1、3、10 mg/kg,口服)或载体(10% DMSO + 90% 生理盐水)处理。在开放式场地箱(40×40×30 cm)中,使用视频追踪软件记录60分钟的运动活动[1] - 条件性回避反应(CAR):雄性Sprague-Dawley大鼠(8-10周龄,每组n=7)在双向穿梭箱中进行训练,通过穿过对面隔间来避免足底电击(0.8 mA)。训练后,在测试前60分钟,大鼠分别接受化合物(1、5、10、30 mg/kg,口服)处理。记录回避率和逃避潜伏期[1] - 新物体识别(PCP模型):小鼠接受PCP(3 mg/kg,腹腔注射)处理以诱导认知功能障碍。30分钟后,给予SEP-363856 HCl(Ulotaront)(5 mg/kg,口服)。训练(接触两个相同的物体)24小时后,对小鼠进行测试,测试内容包括一个熟悉物体和一个新物体,并记录探索时间以计算辨别指数[1] - 僵直测试:小鼠和大鼠分别接受化合物(10、20、30 mg/kg,口服)或氟哌啶醇(1 mg/kg,腹腔注射)处理。通过将动物的前爪放在水平杆(5 cm高)上来评估僵直,并记录移动爪子的潜伏期(最长180秒)[1] - 高架十字迷宫:小鼠在测试前60分钟接受化合物(5 mg/kg,口服)处理。使用视频追踪记录小鼠在开放臂停留的时间和进入开放臂的时间,持续5分钟[1] |
| 药代性质 (ADME/PK) |
口服生物利用度:在 Sprague-Dawley 大鼠中,SEP-363856 HCl(Ulotaront)的口服生物利用度为 78%(10 mg/kg 口服)和 72%(30 mg/kg 口服)[1]
- 血浆药代动力学:口服 10 mg/kg 的大鼠显示 Cmax = 4.2 μM(Tmax = 1 小时),消除半衰期(t1/2)= 7.5 小时,AUC₀-24h = 38.6 μM·h。犬口服 5 mg/kg 后,Cmax = 3.1 μM,t1/2 = 9.2 小时,AUC₀-24h = 29.3 μM·h [1] - 中枢神经系统渗透性:小鼠口服 10 mg/kg 后,给药 2 小时脑组织浓度达到 3.8 μM,脑/血浆浓度比为 0.9 [1] - 代谢:体外肝微粒体试验表明,该药物通过 CYP2D6 和 CYP3A4 代谢。2 小时后,仍有 65% 的母体化合物残留; 50 μM 浓度下未观察到对主要 CYP 同工酶(CYP1A2、2C9、2C19)的抑制作用 [1] - 排泄:在大鼠中,72 小时内 60% 的剂量经粪便排出,30% 经尿液排出,10% 残留在组织中 [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
急性毒性:大鼠单次口服剂量高达 200 mg/kg 的 SEP-363856 HCl(Ulotaront),犬单次口服剂量高达 150 mg/kg,均未出现死亡或急性毒性症状(嗜睡、共济失调、体重减轻)。大鼠 LD50 > 200 mg/kg [1]
- 重复给药毒性:大鼠每日一次口服 10、30 和 100 mg/kg,连续 28 天,血液学(白细胞、红细胞、血小板)或生化(ALT、AST、肌酐、BUN)指标均未见显著变化。脑、肝、肾组织病理学检查未见异常[1] - 血浆蛋白结合率:体外试验表明该化合物与人血浆蛋白的结合率为91%[1] - 无锥体外系反应或升高:大鼠每日口服30 mg/kg,连续14天,未观察到僵直,血浆催乳素水平保持在正常范围内(与氟哌啶醇引起的3倍升高相比)[1] |
| 参考文献 | |
| 其他信息 |
过去50年来,抗精神病药物的临床疗效一直依赖于多巴胺D2受体的阻断。尽管开发非D2化合物旨在避免多巴胺受体阻断带来的副作用,但迄今为止尚未成功研制出新的药物。本研究报道了一种新型精神药物SEP-363856(SEP-856)的发现,该药物具有独特的作用机制。SEP-856的发现源于一项药物化学研究,该研究利用高通量、高内涵的小鼠行为表型分析平台,结合体外筛选,旨在开发非D2(反靶点)化合物,同时使其在多种对D2受体药理机制敏感的动物模型中仍能保持疗效。SEP-856在与精神分裂症相关的啮齿动物模型中表现出广泛的疗效,包括苯环利定(PCP)诱导的活动过度、前脉冲抑制以及PCP诱导的社交互动障碍。除了良好的药代动力学特性、不占据D2受体以及不引起僵直外,SEP-856的广泛疗效还体现在其对大鼠快速眼动睡眠的显著抑制作用上。尽管其作用机制尚未完全阐明,但体外和体内药理学数据以及脑片和体内电生理记录表明,其对痕量胺相关受体1和5-HT1A受体的激动作用是其疗效的关键。基于临床前数据及其独特的作用机制,SEP-856是一种有前景的治疗精神分裂症的新药,代表了一种有望避免因阻断D2信号通路而产生的副作用的新型药物类别。重要性声明:自20世纪50年代氯丙嗪被发现以来,抗精神病药物的临床疗效一直依赖于阻断多巴胺D2受体,但这种方法副作用显著,且对治疗精神分裂症的阴性症状和认知症状疗效甚微。本研究描述了新型精神药物SEP-363856的发现及其药理学特性。SEP-363856并非通过与D2受体直接相互作用发挥其抗精神病样作用。尽管其作用机制尚未完全阐明,但我们的数据表明,其对痕量胺相关受体1和5-HT1A受体的激动作用是其发挥疗效的关键。基于其在临床前动物模型中的独特特性,SEP-363856 有望成为治疗精神分裂症及其他潜在神经精神疾病的候选药物。[1]
背景:SEP-363856 HCl(Ulotaront)是一种新型非典型抗精神病药物,其作用机制与传统的D2受体拮抗剂截然不同。它靶向TAAR1(完全激动剂)和5-HT1A(部分激动剂),满足了精神分裂症治疗中尚未满足的需求(例如,减少锥体外系症状和代谢副作用)[1] - 作用机制:该化合物作为TAAR1的完全激动剂(调节多巴胺和谷氨酸神经传递)和5-HT1A的部分激动剂(调节血清素信号传导)发挥作用。这种双重作用机制可在不阻断D2受体的情况下使精神分裂症患者的异常神经回路恢复正常,从而避免锥体外系反应和血压升高[1] - 治疗潜力:该药物适用于治疗精神分裂症,临床试验表明其能有效减轻阳性症状、阴性症状和认知症状。其良好的安全性(无锥体外系反应,代谢影响极小)支持长期使用[1] - 化学特征:SEP-363856 HCl(优洛酮)是一种小分子药物,分子量约为380 Da(盐酸盐)。它可溶于DMSO(≥ 20 mM)和水溶液(在pH 7.4缓冲液中浓度为2.3 mg/mL),在模拟胃液和肠液中稳定[1] - 临床优势:与典型抗精神病药物(如氟哌啶醇)和某些非典型抗精神病药物(如奥氮平)不同,它不会引起锥体外系症状、高血压或体重增加,从而解决了精神分裂症治疗中的关键副作用问题[1] |
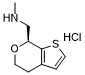
| 分子式 |
C9H14CLNOS
|
|---|---|
| 分子量 |
219.7316
|
| 精确质量 |
219.05
|
| 元素分析 |
C, 49.20; H, 6.42; Cl, 16.13; N, 6.37; O, 7.28; S, 14.59
|
| CAS号 |
1310422-41-3
|
| 相关CAS号 |
SEP-363856; 1310426-33-5; (Rac)-SEP-363856; 1310426-29-9; 2375116-24-6 (besylate); 2375116-27-9 (mesylate)
|
| PubChem CID |
139415154
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
49.5Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
13
|
| 分子复杂度/Complexity |
154
|
| 定义原子立体中心数目 |
1
|
| SMILES |
CNC[C@H]1C2=C(CCO1)C=CS2.Cl
|
| InChi Key |
JRDQGVVFSHRVTL-QRPNPIFTSA-N
|
| InChi Code |
InChI=1S/C9H13NOS.ClH/c1-10-6-8-9-7(2-4-11-8)3-5-12-9;/h3,5,8,10H,2,4,6H2,1H3;1H/t8-;/m0./s1
|
| 化学名 |
1-[(7S)-5,7-dihydro-4H-thieno[2,3-c]pyran-7-yl]-N-methylmethanamine;hydrochloride
|
| 别名 |
Ulotaront; Ulotaront HCl; Ulotaront hydrochloride; SEP 856; SEP-856; SEP856; SEP-363856; SEP 363856; SEP-363856 hydrochloride; SEP-363856 (hydrochloride); SEP-363856 HCl; Ulotaront HCl; ULOTARONT HYDROCHLORIDE; (7S)-4,7-dihydro-N-methyl-5H-thieno[2,3-c]pyran-7-methanamine hydrochloride; 6P8Y2467AU; SEP363856;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O: ~100 mg/mL (~455.1 mM)
DMSO: ~62.5 mg/mL (~284.4 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 6.25 mg/mL (28.44 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 62.5 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 6.25 mg/mL (28.44 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 62.5mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 6.25 mg/mL (28.44 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 100 mg/mL (455.10 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.5510 mL | 22.7552 mL | 45.5104 mL | |
| 5 mM | 0.9102 mL | 4.5510 mL | 9.1021 mL | |
| 10 mM | 0.4551 mL | 2.2755 mL | 4.5510 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04865835 | Completed | Drug: placebo Drug: SEP-363856 Drug: metformin |
Schizophrenia | Sumitomo Pharma America, Inc. | May 12, 2021 | Phase 1 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
