| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 50g |
|
||
| Other Sizes |
|

| 靶点 |
Human Endogenous Metabolite
Uridine (NSC 20256) targets uridine kinase [1] Uridine serves as a substrate for RNA polymerase and DNA polymerase in nucleic acid synthesis [1] - Mitochondrial ATP-dependent potassium channel (mitoKATP) (putative): Uridine's cardioprotective effects are mediated through activation of the mitoKATP channel, as the effect is blocked by the specific inhibitor 5-hydroxydecanoate (5-HD). The mechanism involves its metabolite UDP. However, the provided documents do not include IC50, Ki, or EC50 values for this interaction. [2] - Cellular differentiation/growth regulation: In HL-60 leukemia cells, high concentrations (6-24 mM) of uridine inhibit proliferation and induce differentiation through an unclear mechanism involving intracellular accumulation. [1] |
|---|---|
| 体外研究 (In Vitro) |
促进细胞核酸合成:在人肝细胞系HepG2中,100 μM Uridine处理48小时后,细胞内RNA合成量增加35%,DNA合成量增加28%,为细胞增殖提供必要的核苷原料[1]
- 保护肝细胞免受损伤:50~200 μM Uridine预处理HepG2细胞24小时,可使四氯化碳诱导的细胞凋亡率从42%降至18%,同时增强细胞内谷胱甘肽(GSH)水平,提升抗氧化能力[1] - 支持神经元细胞存活:在原代大鼠皮质神经元细胞中,10 μM Uridine可提高细胞存活率,减少谷氨酸诱导的兴奋性毒性损伤,使坏死细胞比例下降25%[1] - 促进肠道上皮细胞修复:人结肠上皮细胞Caco-2经划伤损伤后,50 μM Uridine处理72小时,细胞迁移率从30%提升至65%,加速创面愈合[1] - HL-60 白血病细胞生长与分化: 尿苷在 6-24 mM 浓度范围内引起时间和浓度依赖性的增殖抑制。在 24 mM 浓度下,它诱导细胞在 G2/M 期积累,并使髓系特异性抗原 Mo 1 上调(第6天约 80% 细胞呈 Mo 1 阳性)。它还能使细胞在 TPA 刺激下快速粘附于塑料表面并伸出长突触,表明分化为混合的粒细胞/巨噬细胞样群体。这些作用可被核苷转运抑制剂 NBMPR(10 μM)部分阻断,并被肌苷(5 mM)拮抗。[1] - 肝细胞保护: 在小鼠 AML12 肝细胞和新鲜分离的大鼠肝细胞中,尿苷(0.2-0.4 mg/mL,预处理 10 小时)显著减轻了 CCl4(10 mM)诱导的细胞毒性。它能减少细胞凋亡(通过 Annexin V-PE/7-AAD 和线粒体膜电位评估),降低 ROS 产生(约降低 2 倍),恢复 S 期细胞比例,并下调 cleaved caspase-3 和 Bax 的表达,同时上调 Bcl-2 的表达。[3] - 肝星状细胞(HSC)活化: 在 TGF-β(25 ng/mL)刺激的 HSC-T6 细胞中,尿苷 处理下调了 α-SMA(HSC 活化标志物)、I 型胶原蛋白和纤连蛋白的表达。Transwell 实验显示,尿苷能抑制活化 HSC 的迁移。[3] |
| 体内研究 (In Vivo) |
小鼠肝损伤模型:腹腔注射Uridine 50 mg/kg,每日1次,连续7天,四氯化碳诱导的肝损伤小鼠血清ALT水平从380 U/L降至120 U/L,AST水平从420 U/L降至150 U/L,肝脏组织炎症浸润减轻[1]
- 大鼠化疗性黏膜炎模型:口服Uridine 100 mg/kg,每日2次,连续5天,氟尿嘧啶诱导的肠道黏膜炎大鼠肠道绒毛高度从200 μm升至350 μm,黏膜损伤评分从7分降至3分[1] - 小鼠神经保护模型:腹腔注射Uridine 30 mg/kg,每日1次,连续14天,可改善东莨菪碱诱导的记忆障碍,Morris水迷宫实验中逃避潜伏期从80秒缩短至45秒[1] - 急性心肌缺血模型(大鼠): 在左冠状动脉(LCA)结扎前 5 分钟静脉注射 尿苷(30 mg/kg)可显著保护心肌。它防止了 ATP 和磷酸肌酸(CrP)水平的下降,减少了脂质过氧化物和共轭二烯,并恢复了超氧化物歧化酶活性和还原型谷胱甘肽水平。这导致缺血改变区和 T 波幅度减小。它还通过减少心律失常总持续时间、室性早搏次数、室性心动过速和心室颤动的持续时间而表现出抗心律失常作用。[2] - 心肌缺血/再灌注模型(大鼠): 尿苷(30 mg/kg,缺血前 30 分钟和再灌注前 5 分钟各给药一次)在再灌注 120 分钟后保留了 ATP 和 CrP 水平,降低了氧化应激标志物,并增加了抗氧化活性。它使梗死面积占风险区的百分比降低了 36%。它减少了 VT 发作的持续时间和次数,但未显著影响再灌注心律失常的发生率。[2] - CCl4 诱导的肝纤维化模型(小鼠): 在 CCl4 诱导 2 周后,通过饮水给予 尿苷(10-20 mg/kg,持续 6 周)。尿苷治疗显著减少了胶原沉积(通过天狼星红和 Masson 染色评估)。纤维化面积从 CCl4 单独组的 18% 降至 13.4%(10 mg/kg)和 8.5%(20 mg/kg)。它还下调了 I 型胶原 mRNA,降低了炎症标志物(TNF-α, IL-1β, MCP-1),抑制了 NF-κB 活化,并减少了肝脏中的 α-SMA 表达。血清肝酶(ALT, AST, ALP)和纤维化标志物(HA, PIIINP)水平也显著降低。[3] |
| 酶活实验 |
- mitoKATP 通道活性(间接): 通过使用特异性 mitoKATP 通道阻滞剂 5-羟基癸酸(5-HD)研究了 尿苷 的心脏保护机制。在 AMI 和 I/R 大鼠模型中,在尿苷给药前 5 分钟预注射 5-HD(5 mg/kg, i.v.)可完全或显著阻断尿苷的节能、抗氧化和抗缺血作用。这间接证实了该通道的参与。该研究未描述对分离通道的直接酶学实验。[2]
|
| 细胞实验 |
- HL-60 细胞增殖与分化实验: 细胞在含 15% FBS 的 RPMI 1640 培养基中培养。使用库尔特计数器每日测量生长。分化通过 NBT 还原实验(TPA 刺激后)和流式细胞术检测 Mo 1 抗体结合来评估。细胞周期分析通过光神霉素染色后的流式细胞术进行。[1]
- 肝细胞活力(MTT)实验: 将新鲜分离的小鼠肝细胞接种于 96 孔板。在存在或不存在尿苷预处理的情况下用 CCl4(10 mM)处理后,加入 MTT 溶液。形成的甲臜晶体溶解于 DMSO,并在 570 nm 处测量吸光度。[3] - 细胞凋亡实验: AML12 肝细胞用 CCl4(10 mM)和/或尿苷处理。使用 Annexin V-PE/7-AAD 凋亡检测试剂盒处理细胞,然后进行流式细胞术以定量细胞凋亡。[3] - 线粒体膜电位实验: 处理后,用 JC-1 探针对 AML12 细胞进行染色。使用共聚焦激光扫描显微镜或流式细胞术分析代表 ΔΨm 的红/绿荧光比值。[3] - 活性氧实验: 使用荧光探针 DCFH-DA(10 μM)测量 AML12 细胞内的 ROS 水平。通过流式细胞术检测荧光,并通过共聚焦显微镜可视化。[3] - HSC 迁移实验(Transwell): 将 TGF-β 活化的 HSC-T6 细胞接种到 Transwell 板的上室,有或无尿苷。下室含 10% FBS 作为化学引诱物。24 小时后,迁移至膜下侧的细胞经固定、苏木精染色和计数。[3] |
| 动物实验 |
- 急性心肌缺血(大鼠): 雄性 Wistar 大鼠(300-350 g)用戊巴比妥钠(50 mg/kg, i.p.)麻醉。左冠状动脉结扎 60 分钟。在结扎前 5 分钟静脉注射 尿苷(30 mg/kg)。mitoKATP 阻滞剂 5-HD(5 mg/kg, i.v.)在尿苷前 5 分钟给予。[2]
- 心肌缺血/再灌注(大鼠): 雄性 Wistar 大鼠进行 30 分钟 LCA 结扎,随后再灌注 120 分钟。尿苷(30 mg/kg, i.v.)给药两次:缺血前 30 分钟和再灌注前 5 分钟。5-HD(5 mg/kg, i.v.)在每次尿苷注射前 5 分钟给予。[2] - CCl4 诱导的肝纤维化(小鼠): 雄性 C57BL/6J 小鼠(19±2 g)腹腔注射溶于橄榄油的 10% CCl4(0.6 μL/g),每周两次,持续 8 周。尿苷 从第 3 周开始通过饮水(10 或 20 mg/kg)给药,持续 6 周。[3] |
| 药代性质 (ADME/PK) |
吸收:口服后在胃肠道迅速吸收;大鼠单次口服 50 mg/kg 后,血浆峰浓度 (Cmax) 为 8 μg/mL,达峰时间 (Tmax) 为 1 小时,口服生物利用度约为 75%[1]
- 分布:广泛分布于全身组织,在肝脏、肾脏、脑和骨骼肌中浓度较高;小鼠静脉注射 30 mg/kg 后,肝脏药物浓度是血浆浓度的 2.5 倍,脑组织药物浓度是血浆浓度的 0.8 倍[1] - 代谢:在细胞内经尿苷激酶磷酸化形成尿苷单磷酸 (UMP),UMP 进一步转化为尿苷二磷酸 (UDP) 和尿苷三磷酸 (UTP),用于核酸合成和能量代谢;在肝脏中经尿苷磷酸化酶部分降解为尿嘧啶和核糖[1] - 排泄:大鼠给药后72小时内,尿液排泄量占给药剂量的60%(主要为代谢物尿嘧啶),粪便排泄量占15%[1] - 半衰期:大鼠静脉注射后消除半衰期(t1/2β)为2.5小时;口服后t1/2β为3.2小时[1] - 血浆蛋白结合率:体外实验表明,该药物在人血浆中的血浆蛋白结合率<10%[1] - 血清消除(大鼠): 在大鼠静脉推注 尿苷(30 mg/kg)后,血清水平在 5 分钟达到峰值(约 65 μmol/L)。在常氧大鼠中,血清水平保持升高超过 65 分钟。在急性心肌缺血大鼠中,尿苷消除更快,注射后 20 分钟内血清水平降至接近背景水平,表明在应激状态下组织消耗增加。基线血清尿苷水平约为 5.9 μmol/L。[2] - 心肌核苷酸水平(大鼠): 在对照大鼠中,给予尿苷(30 mg/kg, i.v.)后 65 分钟,心肌 UDP 水平增加 2.0 倍,UTP 水平增加 2.2 倍。在 AMI 模型中,与未治疗的 AMI 大鼠相比,尿苷治疗显著增加了 UDP 和 UTP 水平,证明外源性尿苷是缺血心肌中核苷酸合成的来源。[2] |
| 毒性/毒理 (Toxicokinetics/TK) |
毒性数据
小鼠(腹腔注射):LD50 4335 mg/kg 急性毒性:小鼠口服半数致死量 (LD50) >5000 mg/kg,静脉注射 LD50 >2000 mg/kg,表明急性毒性极低[1] -慢性毒性:大鼠每日一次口服 500 mg/kg 尿苷,持续 90 天,未见体重增加、血常规或肝肾功能方面的显著异常,组织病理学检查也未发现器官损伤[1] -不良反应:在常规口服剂量(500~1000 mg/天)下,偶见轻微胃肠道不适(恶心、腹胀),发生率 <5%,未见严重不良反应[1] -特殊人群毒性:未见胚胎毒性或在妊娠和哺乳期动物实验中,每日剂量为 100 mg/kg 时观察到了致畸性[1] - 一般毒性: 在动物模型中未报告明显的毒性或显著副作用。在 AMI 和 I/R 研究中,尿苷治疗组的动物存活率与对照组相当。[2] - 肝功能标志物: 在 CCl4 模型中,尿苷 治疗显著降低了升高的血清丙氨酸转氨酶、天冬氨酸转氨酶和碱性磷酸酶水平,表明其对 CCl4 诱导的肝损伤具有保肝作用。对总胆红素和白蛋白无显著影响。[3] - 血流动力学参数: 在大鼠 AMI 和 I/R 模型中,所用剂量的尿苷(30 mg/kg)未引起除缺血诱导的病理改变之外的显著不良血流动力学效应(如血压、心率)。[2] |
| 参考文献 |
[1]. Effects of uridine on the growth and differentiation of HL-60 leukemia cells. Leuk Res. 1991;15(11):1051-8.
[3]. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci Rep. 2021 Aug 20;11(1):16999. [2]. Uridine alleviates carbon tetrachloride-induced liver fibrosis by regulating the activity of liver-related cells. J Cell Mol Med. 2022 Feb;26(3):840-854. |
| 其他信息 |
尿苷是一种核糖核苷,由一个尿嘧啶分子通过β-N(1)-糖苷键与一个核糖呋喃糖部分连接而成。它是一种人体代谢物、基础代谢物和药物代谢物,其功能与尿嘧啶相关。
RG2417 是一种专有的尿苷制剂。尿苷是一种生物活性化合物,对DNA和RNA(存在于所有细胞中的基本遗传物质)的合成以及许多其他对细胞代谢至关重要的因子都至关重要。尿苷由线粒体合成,线粒体是人体细胞的能量工厂,负责能量代谢。临床前和临床研究均支持尿苷疗法在神经精神疾病治疗中的应用。近期报告显示,在双相情感障碍患者的大脑中,编码线粒体蛋白的某些基因显著下调。这项新发现表明,双相情感障碍的症状可能与大脑能量代谢紊乱有关。 尿苷是大肠杆菌(K12菌株、MG1655菌株)中发现或产生的一种代谢产物。 尿苷是一种嘧啶类似物。尿苷的化学分类为嘧啶类化合物及其类似物/衍生物。 据报道,米黑孢霉、地黄以及其他一些有相关数据的生物体中都含有尿苷。 尿苷是一种核苷,由尿嘧啶和D-核糖组成,也是RNA的组成成分。尿苷已被研究作为一种解救剂,用于降低5-氟尿嘧啶(5-FU)的毒性,从而允许在化疗方案中使用更高剂量的5-FU。 (NCI04) 尿苷是酿酒酵母中发现或产生的代谢产物。 它是一种核糖核苷,其中核糖与尿嘧啶相连。 药物适应症 已研究用于治疗双相情感障碍和躁狂症。 背景:尿苷是一种天然存在的嘧啶核苷,广泛存在于动植物细胞中。它是RNA的组成部分,参与DNA合成、能量代谢(UTP作为能量载体)以及糖蛋白和糖脂的合成[1] - 作用机制:作为一种核苷原料,它可以补充体内不足的尿苷,促进核酸合成,并修复受损细胞;它还可通过提高细胞内谷胱甘肽 (GSH) 水平和调节能量代谢发挥细胞保护作用[1] - 适应症相关:用作药物性肝损伤和化疗引起的口腔/肠道黏膜炎的辅助治疗;作为神经退行性疾病(例如阿尔茨海默病)的营养补充剂,以改善认知功能[1] - FDA 状态:被归类为公认安全 (GRAS) 物质,作为膳食补充剂,未获准作为处方药[1] - 心脏保护机制: 尿苷的心脏保护作用被认为是通过其在细胞内转化为 UDP,进而激活 mitoKATP 通道介导的。这有助于在线粒体缺血/再灌注过程中保护线粒体功能、维持 ATP 合成、减少氧化应激和防止细胞死亡。其作用可被特异性 mitoKATP 通道阻滞剂 5-HD 阻断。[2] - 抗纤维化机制: 尿苷通过多种途径缓解 CCl4 诱导的肝纤维化:(1) 保护肝细胞免受凋亡和氧化应激;(2) 减少炎症细胞因子(TNF-α, IL-1β, MCP-1)的表达并抑制 NF-κB 活化;(3) 下调活化的肝星状细胞中 α-SMA、I 型胶原蛋白和纤连蛋白的表达,并抑制其迁移。[3] - 对 HL-60 分化的影响: 高浓度的尿苷(24 mM)可强制 HL-60 白血病细胞分化,提示其在分化治疗中的潜在作用,但所需的高浓度限制了其临床相关性。该效应与分化过程中诱导 Na+ 依赖性核苷转运系统后的细胞内尿苷蓄积有关。[1] |
| 分子式 |
C9H12N2O6
|
|
|---|---|---|
| 分子量 |
244.2
|
|
| 精确质量 |
244.069
|
|
| 元素分析 |
C, 44.27; H, 4.95; N, 11.47; O, 39.31
|
|
| CAS号 |
58-96-8
|
|
| 相关CAS号 |
|
|
| PubChem CID |
6029
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.9±0.1 g/cm3
|
|
| 沸点 |
567.9±60.0 °C at 760 mmHg
|
|
| 熔点 |
163 °C
|
|
| 闪点 |
297.2±32.9 °C
|
|
| 蒸汽压 |
0.0±3.5 mmHg at 25°C
|
|
| 折射率 |
1.732
|
|
| LogP |
-1.55
|
|
| tPSA |
124.78
|
|
| 氢键供体(HBD)数目 |
4
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
17
|
|
| 分子复杂度/Complexity |
371
|
|
| 定义原子立体中心数目 |
4
|
|
| SMILES |
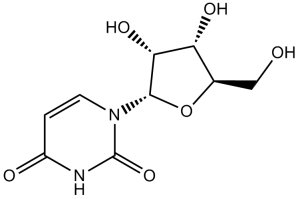
O1[C@]([H])(C([H])([H])O[H])[C@]([H])([C@]([H])([C@]1([H])N1C([H])=C([H])C(N([H])C1=O)=O)O[H])O[H]
|
|
| InChi Key |
DRTQHJPVMGBUCF-XVFCMESISA-N
|
|
| InChi Code |
InChI=1S/C9H12N2O6/c12-3-4-6(14)7(15)8(17-4)11-2-1-5(13)10-9(11)16/h1-2,4,6-8,12,14-15H,3H2,(H,10,13,16)/t4-,6-,7-,8-/m1/s1
|
|
| 化学名 |
1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione
|
|
| 别名 |
uridine; 58-96-8; Uridin; Uracil riboside; 1-beta-D-Ribofuranosyluracil; NSC 20256; NSC-20256; NSC20256
|
|
| HS Tariff Code |
2934.99.03.00
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (10.24 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (10.24 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (10.24 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 40 mg/mL (163.80 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0950 mL | 20.4750 mL | 40.9500 mL | |
| 5 mM | 0.8190 mL | 4.0950 mL | 8.1900 mL | |
| 10 mM | 0.4095 mL | 2.0475 mL | 4.0950 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT03265964
Conditions:Suicidal IdeationLink: https://clinicaltrials.gov/ct2/show/NCT00841269
Conditions:Bipolar DisorderLink: https://clinicaltrials.gov/ct2/show/NCT01805440
Conditions:Bipolar Disorder|Bipolar Depression|Manic Depression
Title:Immunologic Effects of Supplemental Monosaccharide and Nucleoside Derivatives in Patients With Inherited Disorders of Glycosylation
Status:Terminated
updateDate:2017-07-02
Ctid:NCT02511041
Link: https://clinicaltrials.gov/ct2/show/NCT02511041
Conditions:PGM3Link: https://clinicaltrials.gov/ct2/show/NCT00004705
Conditions:Cystic FibrosisLink: https://clinicaltrials.gov/ct2/show/NCT00812058
Conditions:Bipolar I DepressionLink: https://clinicaltrials.gov/ct2/show/NCT01261260
Conditions:Healthy Male SubjectsLink: https://clinicaltrials.gov/ct2/show/NCT00322764
Conditions:Bipolar DepressionLink: https://clinicaltrials.gov/ct2/show/NCT00004658
Conditions:5'-Nucleotidase Syndrome (S,S)-D211
(S,S)-D211
 Di(MMY-SJG)-Ph-prop-2-yne-1-sulfonic acid
Di(MMY-SJG)-Ph-prop-2-yne-1-sulfonic acid
 MOMA-341
MOMA-341
 Morpholino G phosphoramidite
Morpholino G phosphoramidite
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
