| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 500μg |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
Quinolone antibiotic; DNA gyrase/topoisomerase
Zoliflodacin (formerly ETX-0914; AZD-0914; ETX0914; AZD0914) possesses antibacterial activity against important fastidious Gram-negative (Haemophilus influenzae, Neisseria gonorrhoeae), atypical (Legionella pneumophila), anaerobic (Clostridium difficile), and Gram-positive (Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus pneumoniae, Streptococcus pyogenes, and Streptococcus agalactiae), fastidious Gram-negative (Haemophilus influenzae, Neisseria gonorrhoeae), and atypical (Legionella pneumophila) bacterial species, including isolates with documented fluoroquinolone resistance. The mechanism of action of Zoliflodacin is demonstrated to be different from that of other commercially available antibacterial compounds, such as fluoroquinolones, in that it inhibits DNA biosynthesis and accumulates double-strand cleavages, thereby exerting antibacterial activity. Under permissive conditions, zoliflodacin stabilizes and arrests the cleaved covalent complex of gyrase with double-strand broken DNA, preventing the double-strand cleaved DNA from religating to form fused circular DNA[1]. |
|---|---|
| 体外研究 (In Vitro) |
Zoliflodacin(以前的 ETX-0914;AZD-0914;ETX0914;AZD0914)对关键革兰氏阳性菌(金黄色葡萄球菌、表皮葡萄球菌、肺炎链球菌、化脓性链球菌和无乳链球菌)、挑剔的革兰氏阴性菌(流感嗜血杆菌、奈瑟菌)具有抗菌活性。淋病菌)、非典型菌(嗜肺军团菌)和厌氧菌(艰难梭菌),包括已知对氟喹诺酮类药物耐药的分离株。 Zoliflodacin 的抗菌活性是通过抑制 DNA 生物合成和双链裂解的积累来实现的;这种作用机制不同于其他市售抗菌化合物,包括氟喹诺酮类药物。在允许的条件下,Zoliflodacin 可稳定并阻止旋转酶与双链断裂 DNA 的切割共价复合物,从而阻止双链切割 DNA 的再连接形成融合的环状 DNA[1]。
Zoliflodacin (AZD0914) 在体外对多种革兰氏阳性菌、苛养革兰氏阴性菌、非典型细菌和厌氧菌(包括耐药菌株)表现出强大的抗菌活性。 针对100株金黄色葡萄球菌(包括MRSA),MIC范围为0.06–0.5 µg/mL,MIC₅₀和MIC₉₀均为0.25 µg/mL。其对左氧氟沙星耐药(MIC₉₀: >16 µg/mL)、万古霉素中介(VISA)、万古霉素耐药(VRSA)和利奈唑胺耐药的金黄色葡萄球菌分离株仍保持活性。[1] 针对98株肺炎链球菌,MIC范围为0.06–0.5 µg/mL,MIC₅₀和MIC₉₀分别为0.125和0.25 µg/mL。其对左氧氟沙星耐药和大环内酯类耐药的分离株仍保持活性。[1] 针对37株淋病奈瑟菌,MIC范围为0.03–0.25 µg/mL,MIC₅₀和MIC₉₀分别为0.06和0.125 µg/mL。其对环丙沙星耐药(MIC范围:0.03–0.125 µg/mL)以及一株头孢曲松和环丙沙星双重耐药的分离株(MIC:0.06 µg/mL)仍保持活性。[1] 针对29株流感嗜血杆菌,MIC范围为0.125–1 µg/mL,MIC₅₀和MIC₉₀分别为0.25和1 µg/mL。[1] 针对10株嗜肺军团菌,MIC范围为0.008–0.06 µg/mL,MIC₅₀和MIC₉₀分别为0.03和0.06 µg/mL。[1] 针对2株艰难梭菌,MIC为0.125 µg/mL。[1] 其对非苛养革兰氏阴性病原体(如大肠杆菌、肺炎克雷伯菌、铜绿假单胞菌和鲍曼不动杆菌)的活性较低(MIC₉₀范围为4至>128 µg/mL)。外排泵(如大肠杆菌中的tolC、铜绿假单胞菌中的mexABCDXY)是导致其对革兰氏阴性菌活性降低的因素之一。[1] 针对7株金黄色葡萄球菌和1株化脓性链球菌的最低杀菌浓度测试表明,Zoliflodacin (AZD0914) 具有杀菌作用,MBC值在MIC的2倍范围内。[1] 针对左氧氟沙星耐药的MRSA(USA100)和肺炎链球菌ATCC 49619的体外时间-杀菌曲线研究显示,在≥2倍MIC的浓度下,6小时内CFU/mL降低≥3 log₁₀。[1] Zoliflodacin (AZD0914) 对USA100和肺炎链球菌ATCC 49619的抗生素后效应(PAE)范围为0.25至>3.3小时,并随浓度增加而延长。[1] 金黄色葡萄球菌对AZD0914的自发耐药频率较低(在4-8倍MIC下为10⁻⁹至<10⁻¹⁰),与利奈唑胺相当,低于左氧氟沙星。获得的耐药突变定位于gyrB,其MIC增加≥4倍。[1] 未观察到与已知的导致对氟喹诺酮类、新生霉素或香豆霉素耐药的gyrA、gyrB、parC或parE突变菌株的交叉耐药。与野生型菌株相比,AZD0914的MIC变化不超过2倍。[1] 在与17种对照抗菌药物对5种参考菌株的棋盘法协同试验中,仅观察到相加/无关作用(平均FICI:0.77–1.66),未观察到协同或拮抗作用。[1] |
| 体内研究 (In Vivo) |
美国疾病控制中心和世界卫生组织发布了一份优先病原体清单,其中治疗选择正在减少,包括抗生素耐药性淋病奈瑟菌,迫切需要新型口服药物。Zoliflodacin是一种名为螺嘧啶三酮的新型抗菌剂中的第一种,正在开发用于治疗淋病。它具有独特的抑制细菌II型拓扑异构酶的模式,细菌回旋酶中的结合位点不同于氟喹诺酮类药物。Zoliflodacin具有杀菌作用,对淋病奈瑟菌具有较低的耐药频率和强效抗菌活性,包括多重耐药菌株(MIC范围≤0.002至0.25μg/mL)。尽管唑氟达秦是为治疗淋病而开发的,但它对革兰氏阳性、挑剔的革兰氏阴性和非典型病原体也有活性。使用金黄色葡萄球菌的中空纤维感染模型表明,fAUC/MIC的药代动力学/药效学指数与小鼠体内中性粒细胞减少性大腿模型的疗效相关性最好。随后,这些数据和来自大腿模型的未结合暴露量被用于替代病原体方法,以确定淋病奈瑟菌临床开发的剂量范围。在临床前研究中,广泛的安全裕度支持在健康志愿者中进行1期研究,该研究显示了线性药代动力学、良好的口服生物利用度,并且没有显著的安全性发现。在一项2期研究中,唑氟达秦对治疗淋球菌泌尿生殖道和直肠感染有效。与全球抗生素研究发展计划(GARDP)合作,唑氟达新目前正在全球3期临床试验中进行研究。Zoliflodacin代表了一种有前景的新型口服疗法,用于治疗由淋病奈瑟菌引起的耐药性感染[2]。
|
| 酶活实验 |
AZD0914是一种新型的螺旋嘧啶三酮细菌DNA旋转酶/拓扑异构酶抑制剂,对关键的革兰氏阳性(金黄色葡萄球菌、表皮葡萄球菌、肺炎链球菌、化脓性链球菌和无乳链球菌)、挑剔的革兰氏阴性(流感嗜血杆菌和淋病奈瑟菌)、非典型(嗜肺军团菌)和厌氧(艰难梭菌)细菌种类具有强效的体外抗菌活性,包括已知对氟喹诺酮类药物有耐药性的分离株。AZD0914通过抑制DNA生物合成和双链裂解的积累起作用;这种抑制机制不同于其他市场上销售的抗菌化合物。AZD0914在允许的条件下稳定并阻止回转酶与双链断裂DNA的切割共价复合物,从而阻止双链切割DNA的再结合形成融合的环状DNA。尽管这种机制与氟喹诺酮类药物相似,但在机制上是不同的。AZD0914在金黄色葡萄球菌中表现出较低的自发耐药性,如果获得突变体,则突变定位于gyrB。此外,AZD0914对最近对氟喹诺酮类或其他药物类别(包括大环内酯类、β-内酰胺类、糖肽和恶唑烷酮)产生耐药性的细菌临床分离株没有观察到交叉耐药性。AZD0914在最低杀菌浓度和体外时间杀灭研究中都具有杀菌作用。在17种对比抗菌药物的体外棋盘格/协同试验中,仅观察到加性/无差异性。AZD0914的强效体外抗菌活性(包括对氟喹诺酮耐药分离株的活性)、低耐药频率、缺乏交叉耐药和杀菌活性支持其持续发展[1]。
|
| 细胞实验 |
测定 Zoliflodacin(以前的 ETX-0914;AZD-0914;ETX0914;AZD0914)肉汤宏观稀释 MIC,并将其用作体外时间杀灭和抗生素后效应 (PAE) 测试的起点。使用含有 10 mL 体积的阳离子调节 Mueller-Hinton 肉汤的玻璃管(18 x 150 mm,无搅拌)进行体外静态时间杀灭研究,培养物按对数生长(起始接种量为 1×106 CFU/mL)。左氧氟沙星敏感和左氧氟沙星耐药的金黄色葡萄球菌。唑利氟达星的测试浓度相当于 MIC 的 0.5、1、2、4 和 8 倍;如前所述,将 100 μL 等份样品点样到 25 ml 羊血琼脂平板上,在 0、2、4、6、8 和 24 小时对样品进行平板接种以进行菌落计数。化合物在最低药物浓度下被认为具有杀菌作用,可在 24 小时内将活生物体计数减少 ≥3 log10。节省时间的研究一式两份进行;组合测试并报告平均值[1]。
体外时间-杀菌实验在装有阳离子调节Mueller-Hinton肉汤的玻璃管中进行。将处于对数生长期的金黄色葡萄球菌(USA300和USA100)或肺炎链球菌(ATCC 49619)起始接种量调整为1 × 10⁶ CFU/mL,并暴露于相当于0.5、1、2、4和8倍MIC浓度的Zoliflodacin (AZD0914)。在0、2、4、6、8和24小时取样,将100 µL等分试样点种子绵羊血琼脂平板上进行菌落计数。杀菌活性定义为24小时内活菌数减少≥3 log₁₀。实验重复进行两次,报告平均值。[1] 抗生素后效应(PAE)测试方法类似。将培养物暴露于0.5、1、4或16倍MIC的Zoliflodacin (AZD0914) 中1小时。然后通过1:1000稀释去除药物。PAE定义为药物暴露组和未暴露组培养物在稀释后活菌数增加1 log₁₀ CFU/mL所需时间的差值。[1] |
| 动物实验 |
直到最近,由于缺乏可靠的淋病奈瑟菌动物模型,人们对该病原体的药代动力学/药效学(PK/PD)和暴露靶点的理解一直受到阻碍。由于宿主系统内淋病奈瑟菌的自发清除,成功定植往往难以实现,且难以在治疗窗口内维持,这限制了该模型在评估新型抗生素方面的应用。体外动态模型系统,例如用于确定PK/PD驱动因素和评估临床剂量方案的中空纤维模型,直到最近才在淋病奈瑟菌模型中得到成功应用。目前临床上使用的疗法和方案通常依赖于现有药物临床应用提供的轶事证据,这些证据表明宿主对该病原体具有很强的敏感性。由于佐利氟达星的作用机制与其他已成功用于治疗淋病的上市拓扑异构酶抑制剂相似,因此采用金黄色葡萄球菌作为替代病原体来研究佐利氟达星。实验得出的PK/PD靶点转化是基于以下前提:从鼠模型参考靶点获得的游离血浆靶点与临床疗效相关的靶点相匹配。尽管拓扑异构酶抑制剂相关的PK/PD驱动因素历来是浓度-时间曲线下面积(AUC)/最小抑菌浓度(MIC),但一系列体外空心纤维实验证实了佐利氟达星本身也具有这种相关性。在这些实验中,模拟了游离药物浓度-时间曲线,并进行剂量分割,以区分AUC/MIC、Cmax/AUC和T > MIC这三种PK/PD驱动因素。本研究采用甲氧西林敏感金黄色葡萄球菌菌株(ARC516)进行试验,结果表明AUC/MIC是与佐利氟达星活性最密切相关的指标,[R²] = 0.95(图4)。[2]
在空心纤维模型中确定佐利氟达星对金黄色葡萄球菌的药代动力学/药效学驱动因素后,本研究采用甲氧西林敏感金黄色葡萄球菌菌株(ARC516)建立小鼠大腿感染模型,分别在免疫功能正常和免疫抑制小鼠中评估佐利氟达星的体内疗效。佐利氟达星在两种小鼠模型中均表现出剂量依赖性的高效性。当AUC为20 μg·h/mL时即可达到抑菌效果。考虑到佐利氟达星在小鼠体内的蛋白结合率(81%),中性粒细胞减少动物的菌落形成单位/克(CFU/g)最大下降3个对数单位(log10)对应于游离药物AUC为23 μg·h/mL。本研究使用的金黄色葡萄球菌ARC516对佐利氟达星的MIC为0.0625 μg/mL,菌落数下降超过3个对数单位时,fAUC/MIC为368。为了确定与菌落抑制和菌落数下降1个对数单位(log10)相关的平均游离药物暴露量,本研究在中性粒细胞减少的大腿感染模型中对其他金黄色葡萄球菌菌株(包括耐甲氧西林金黄色葡萄球菌ATCC 33591、USA100 NRS382和USA300 NR538)进行了体内研究。根据合并的体内数据集,与菌落抑制和菌落数下降1个对数单位(log10)相关的fAUC/MIC值分别为66和139。这些数值最终被用作基于药代动力学/药效学(PK/PD)的暴露目标值,用于临床剂量论证。随后,利用中空纤维中唑利氟达星对金黄色葡萄球菌的PK/PD指标fAUC/MIC以及在小鼠体内中性粒细胞减少性大腿模型中获得的游离暴露量,采用替代病原体方法,帮助确定淋病奈瑟菌临床开发的剂量范围。为了支持该方法,我们获得了氧氟沙星和环丙沙星作为参考化合物,用于治疗淋病奈瑟菌和金黄色葡萄球菌引起的皮肤感染的临床剂量对应的fAUC/MIC值,如表6所示。[2] |
| 药代性质 (ADME/PK) |
吸收
空腹状态下,血浆药物浓度达峰时间(Tmax)中位数为2.5小时;餐后状态下为4.0小时。食物摄入显著影响吸收——与空腹状态相比,与中等或高脂肪食物同服可使Cmax增加约1.5倍,AUC增加约1.5至2倍。在接受3克剂量治疗的单纯性泌尿生殖道淋病患者中,Cmax的几何平均值为28.5微克/毫升。 消除途径 佐利氟达星的主要消除途径是粪便。约79.6%的药物相关物质从粪便中排出(其中1.5%为原药),18.2%从尿液中排出(其中2.5%为原药)。 分布容积 佐利氟达辛的几何平均表观分布容积在空腹状态下为 177 L,在进食状态下为 98.7 L。 清除率 佐利氟达辛的几何平均表观全身清除率在空腹状态下为 19.1 L/h,在进食状态下为 12.5 L/h。 蛋白结合率 佐利氟达辛与人血浆蛋白的结合率为 83%。 代谢/代谢物 佐利氟达辛的主要清除机制是通过 CYP 介导和非 CYP 介导的代谢途径,但非 CYP 代谢途径尚未完全阐明。 CYP表型研究表明,CYP介导的代谢主要通过CYP3A4/5(68%)进行,CYP1A2(14%)、CYP2C9(10%)、CYP2C8(5%)和CYP2C19(2.7%)的贡献较小。 生物半衰期 佐利氟达辛的几何平均消除半衰期在空腹状态下为6.4小时,在进食状态下为5.5小时。 |
| 参考文献 |
|
| 其他信息 |
佐利氟达辛已用于淋病基础科学和治疗研究的临床试验。
为了应对多重耐药淋病奈瑟菌带来的未满足的医疗需求,佐利氟达辛(曾用名AZD0914、ETX0914)作为一种新型抗菌药物——螺嘧啶三酮类药物,被开发用于治疗非复杂性淋病。佐利氟达辛是一种口服生物利用度高的抗菌药物,其杀菌机制是通过抑制细菌II型拓扑异构酶。本综述介绍了该新型口服抗生素的临床前和早期临床数据。[2] 佐利氟达辛是一种新型螺嘧啶三酮类抗生素,由Entasis Therapeutics公司开发,用于治疗非复杂性淋病,是首个新型拓扑异构酶抑制剂类药物。由于其独特的作用机制,佐利氟达星与氟喹诺酮类药物不存在交叉耐药性,因此可能有效治疗由氟喹诺酮类耐药淋病奈瑟菌菌株引起的感染。一项II期研究证实了其安全性和有效性,支持了这一观点。Entasis公司已与全球抗生素研发计划(GARDP)合作,继续推进佐利氟达星的研发,并于近期宣布启动一项全球III期研究(https://gardp.org/news-resources/gardp-and-entasis-therapeutics-initiate-global-phase-3-trial-of-zoliflodacin-a-first-in-class-oral-antibiotic-for-the-treatment-of-gonorrhoea/)。这是一项开放标签试验,比较单次口服3克佐利氟达星与头孢曲松联合阿奇霉素的疗效(2:1随机分组)。与二期研究类似,三期试验也以淋病奈瑟菌引起的泌尿生殖系统感染作为入组标准。然而,本试验还将泌尿生殖系统外感染作为次要终点进行研究,希望这些数据也能帮助我们了解佐利氟达星治疗这些感染的疗效。这项临床研究预计将纳入来自美国、荷兰、泰国和南非的约 1000 名患有泌尿生殖道淋病的成年患者(ClinicalTrials.gov 注册号:NCT03959527),预计结果将于 2021 年公布。进入 2020 年代,如果该试验取得积极结果,则表明佐利氟达星可作为口服单药疗法用于治疗淋病。[2] 佐利氟达星 (AZD0914) 是一种新型的口服活性螺嘧啶三酮类抗生素,旨在治疗由耐药革兰氏阳性菌和难培养革兰氏阴性菌引起的感染,包括耐甲氧西林金黄色葡萄球菌 (MRSA)、耐药肺炎链球菌和多重耐药淋病奈瑟菌。 [1] 其作用机制涉及通过稳定裂解的DNA促旋酶-DNA复合物来抑制DNA生物合成,这与氟喹诺酮类药物的作用机制截然不同。[1] 该化合物对氟喹诺酮类耐药菌株和头孢曲松耐药菌株具有强效活性,耐药率低,无交叉耐药性,且具有杀菌特性,这些都支持其继续研发。[1] 该化合物已获得合格传染病产品(QIDP)认定和快速通道资格,目前正在评估其治疗由淋病奈瑟菌引起的性传播感染的疗效。[1] |
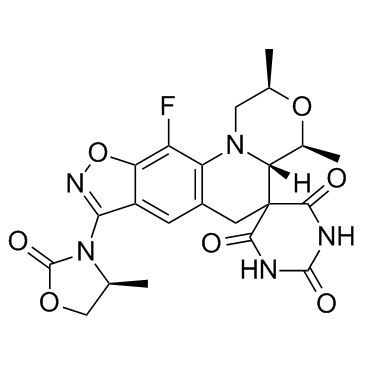
| 分子式 |
C22H22N5O7F
|
|---|---|
| 分子量 |
487.438
|
| 精确质量 |
487.15
|
| 元素分析 |
C, 54.21; H, 4.55; F, 3.90; N, 14.37; O, 22.98
|
| CAS号 |
1620458-09-4
|
| PubChem CID |
76685216
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.6±0.1 g/cm3
|
| 折射率 |
1.685
|
| LogP |
1.77
|
| tPSA |
143Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
10
|
| 可旋转键数目(RBC) |
1
|
| 重原子数目 |
35
|
| 分子复杂度/Complexity |
962
|
| 定义原子立体中心数目 |
4
|
| SMILES |
O=C(NC(C12[C@]([C@H](C)O[C@H](C)C3)([H])N3C4=C(C=C5C(ON=C5N6C(OC[C@@H]6C)=O)=C4F)C2)=O)NC1=O
|
| InChi Key |
ZSWMIFNWDQEXDT-ZESJGQACSA-N
|
| InChi Code |
InChI=1S/C22H22FN5O7/c1-8-7-33-21(32)28(8)17-12-4-11-5-22(18(29)24-20(31)25-19(22)30)16-10(3)34-9(2)6-27(16)14(11)13(23)15(12)35-26-17/h4,8-10,16H,5-7H2,1-3H3,(H2,24,25,29,30,31)/t8-,9+,10-,16+/m0/s1
|
| 化学名 |
(4'R,6'S,7'S)-17'-fluoro-4',6'-dimethyl-13'-[(4S)-4-methyl-2-oxo-1,3-oxazolidin-3-yl]spiro[1,3-diazinane-5,8'-5,15-dioxa-2,14-diazatetracyclo[8.7.0.02,7.012,16]heptadeca-1(17),10,12(16),13-tetraene]-2,4,6-trione
|
| 别名 |
Zoliflodacin; ETX0914; AZD-0914; ETX-0914; AZD0914; ETX 0914; AZD 0914; AZD0914; AZD-0914; Zoliflodacin [USAN]; FWL2263R77;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~140 mg/mL (~287.2 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.27 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.27 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (4.27 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0515 mL | 10.2577 mL | 20.5153 mL | |
| 5 mM | 0.4103 mL | 2.0515 mL | 4.1031 mL | |
| 10 mM | 0.2052 mL | 1.0258 mL | 2.0515 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03959527 | Active Recruiting |
Drug: zoliflodacin Drug: ceftriaxone |
Gonorrhea | Global Antibiotics Research and Development Partnership |
November 6, 2019 | Phase 3 |
| NCT03404167 | Completed | Drug: AZD0914 | Gonorrhoea | National Institute of Allergy and Infectious Diseases (NIAID) |
February 2, 2018 | Phase 1 |
| NCT03718806 | Completed | Other: high calorie, high fat breakfast Drug: Zoliflodacin |
Gonorrhea | Drugs for Neglected Diseases | October 3, 2018 | Phase 1 |
| NCT05635305 | Completed | Drug: Zoliflodacin Patheon | Healthy Volunteers | Global Antibiotics Research and Development Partnership |
November 9, 2022 | Phase 1 |
| NCT02257918 | Completed | Drug: AZD0914 Drug: Ceftriaxone |
Gonorrhoea | National Institute of Allergy and Infectious Diseases (NIAID) |
November 25, 2014 | Phase 2 |
|
In vitrotime-kill performance of AZD0914 (MIC, 0.25 μg/ml) against S. pneumoniae ATCC 49619.Antimicrob Agents Chemother. 2015 Jan; 59(1): 467–474. |
PAE of AZD0914 (MIC, 0.25 μg/ml) againstS. pneumoniaeATCC 49619.Antimicrob Agents Chemother. 2015 Jan; 59(1): 467–474. |
 (S,S)-D211
(S,S)-D211
 Di(MMY-SJG)-Ph-prop-2-yne-1-sulfonic acid
Di(MMY-SJG)-Ph-prop-2-yne-1-sulfonic acid
 MOMA-341
MOMA-341
 Morpholino G phosphoramidite
Morpholino G phosphoramidite
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
