| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
ARN272 (CAS#: 488793-85-7) targets the anandamide transport mechanism, specifically FLAT (FAAH-1-like anandamide transporter). No IC₅₀, Ki, EC₅₀, or DC₅₀ values are reported in the provided literature.[1]
The anti-nausea and anti-emetic effects of ARN272 (CAS#: 488793-85-7) are mediated via indirect activation of cannabinoid CB₁ receptors, as shown by reversal with the CB₁ antagonist SR141716.[1] |
|---|---|
| 体外研究 (In Vitro) |
所提供的文献中没有报告体外活性数据。
|
| 体内研究 (In Vivo) |
采用全身给药花生四烯酸乙醇胺转运抑制剂ARN272([(4-(5-(4-羟基苯基)-3,4-二氮杂双环[4.4.0]癸-1(6),2,4,7,9-戊烯-2-基氨基)-苯基)-苯基氨基-甲酮])来评估预防大鼠氯化锂诱导的恶心行为(条件性张口)和鼩鼱(Suncus murinus)氯化锂诱导的呕吐。本研究通过全身联合给药SR141716拮抗大麻素1 (CB1)受体,探讨了延长内源性大麻素(anandamide)的可用性如何调节大鼠的恶心反应。全身给药ARN272可剂量依赖性地抑制大鼠恶心诱导的条件性张口反应,并剂量依赖性地减少鼩鼱的呕吐。在大鼠中,全身联合给药SR141716和ARN272(剂量为3.0 mg·kg⁻¹)可完全逆转ARN272(剂量为1.0 mg·kg⁻¹)对张口反应的抑制作用。单独使用SR141716与溶剂对照组无显著差异。这些结果表明,化合物ARN272对花生四烯酸乙醇胺(anandamide)转运的抑制作用持续激活CB1受体,从而产生一种间接激动作用,调节毒素引起的恶心和呕吐。这些结果还提供了行为学证据,支持花生四烯酸乙醇胺细胞重摄取过程中采用的易化转运机制。[1]
在大鼠中,腹腔注射ARN272(CAS#: 488793-85-7)(0.1、1.0和3.0 mg/kg)可剂量依赖性地抑制氯化锂(LiCl)诱导的条件性张口(恶心行为)。最高剂量(3.0 mg/kg)与溶剂对照组相比,张口次数显著减少(P < 0.001)。 CB₁受体拮抗剂SR141716(1.0 mg/kg,腹腔注射)与ARN272(3.0 mg/kg)联合给药可完全逆转ARN272的止吐作用,而单独使用SR141716则无此作用。ARN272并未减弱条件性味觉回避(CTA),表明其作用机制为选择性抑制恶心而非影响学习/记忆。[1] 在麝鼩(Suncus murinus)中,全身性给予ARN272(CAS#: 488793-85-7)(9.0和18.0 mg/kg,腹腔注射)可剂量依赖性地降低氯化锂(LiCl)诱导的呕吐。与载体对照组相比,18.0 mg/kg 剂量显著减少了呕吐次数(P < 0.05)。[1] |
| 细胞实验 |
总共 58 只幼稚雄性 Sprague-Dawley 大鼠用于评估抗恶心行为。大鼠被单独饲养在 48 × 26 × 20 厘米的鞋盒笼中,笼子位于群居室,环境温度为 21°C。所有动物都处于反向明暗循环(07:00 关灯;19:00 开灯),可自由进食(Iams 啮齿动物饲料,18% 蛋白质)和自来水,测试期间除外,测试发生在黑暗周期。所有动物都获得了丰富的环境,包括两张干净的纸巾(每周在换笼时补充)和一个 14 厘米长、12 厘米直径的软塑料容器。
总共 21 只 S. murinus(麝鼩)在圭尔夫大学的一个群居中繁殖和饲养。将鼩鼱单独饲养在笼子中,环境温度为 21°C,光照时间为 14/10(晚上 21 点关灯)。在鼩鼱的光照周期内进行测试,时间为 09:00 至 14:00。实验中使用了雄性(42.9–53.0 克)和雌性(26.1–32.9 克)并均匀分布在各组中,实验对象年龄从 98 至 814 天不等。在任何分析中,两性的呕吐频率均无显著差异;因此,在所有报告的分析中,雄性和雌性都被合并在一起。鼩鼱之前有过呕吐经历,每次治疗之间至少需要 3 周的恢复时间。 所有实验中,anandamide 转运抑制剂 ARN272 均在 1:1:8 PEG400、Tween 80 和生理盐水的载体溶液 (VEH) 中制备。所有全身注射均通过腹腔注射给药。在实验 1 中,将 ARN272 以 0.1 mg·mL−1(0.1 mg·kg−1 剂量)、1 mg·mL−1(1 mg·kg−1 剂量)和 3 mg·mL−1(3 mg·kg−1 剂量)的浓度以及 1 mL·kg−1 的体积输送给大鼠,此剂量选择基于 Fu 等人 (2012) 进行的先前实验,其中 1 mg·kg−1 在给药后 2 小时增加血浆中 anandamide 的水平。在实验 2 中,将 ARN272 浓度为 3.0 mg·mL−1(9.0 mg·kg−1 剂量)的剂量以 3 和 3.0 mg·mL−1(18 mg·kg−1 剂量)的剂量以 6 mL·kg−1 的体积输送给鼩鼱,选择浓度和体积的依据是,在之前的实验中,S. murinus 需要的剂量至少增加到大鼠有效剂量的三倍。实验 1 中 SR141716 的浓度为 1.0 mg·mL−1(1.0 mg·kg−1 剂量),体积为 1 mL·kg−1。选择 1.0 mg·kg−1 SR141716 的剂量是基于其在逆转断点和恢复尼古丁自我给药方面的先前有效性,同时还发现与 2.5 mg·kg−1 或更高剂量的 SR141716 不同,它不会增强催吐剂的效果。 所有实验中使用的 0.15 M LiCl 药物治疗均在无菌水中制备,并在实验 1 中以 20 mL·kg−1(127 mg·kg−1)的体积给大鼠给药,在实验 2 中以 60 mL kg−1(390 mg·kg−1)的体积给鼩鼱给药。 |
| 动物实验 |
共使用了58只未经训练的雄性Sprague-Dawley大鼠进行抗恶心行为评估。大鼠单独饲养于48 × 26 × 20 cm的鞋盒式笼中,置于温度为21°C的饲养室中。所有动物均采用反向光暗循环(7:00关灯,19:00开灯),并可自由获取食物(Iams啮齿动物饲料,18%蛋白质)和自来水,测试期间除外(测试在黑暗期进行)。所有动物均通过两张干净的纸巾(每周换笼时补充)和一个长14 cm、直径12 cm的软塑料容器进行环境丰富化。
共饲养了21只家麝鼩(S. murinus),它们在圭尔夫大学的饲养室中繁殖和饲养。鼩鼱单独饲养于笼中,环境温度为21°C,光照周期为14/10小时(21:00关灯)。鼩鼱在光照周期内进行测试,时间为09:00至14:00。实验采用雄性(42.9–53.0 g)和雌性(26.1–32.9 g)鼩鼱,并平均分配到各组,受试者年龄范围为98至814天。所有分析均显示,雄性和雌性鼩鼱的呕吐频率无显著差异;因此,所有报告的分析均将雄性和雌性鼩鼱合并。这些鼩鼱之前接受过催吐剂治疗,但两次治疗之间至少间隔3周的恢复期。 所有实验均使用花生四烯酸乙醇胺转运抑制剂ARN272配制于由PEG400、Tween 80和生理盐水按1:1:8比例混合而成的溶剂(VEH)中。所有全身注射均采用腹腔注射。在实验 1 中,ARN272 以 0.1 mg·mL−1(0.1 mg·kg−1 剂量)、1 mg·mL−1(1 mg·kg−1 剂量)和 3 mg·mL−1(3 mg·kg−1 剂量)的浓度,以及 1 mL·kg−1 的体积,被给予大鼠。这是根据 Fu 等人 (2012) 先前进行的实验选择的,其中 1 mg·kg−1 的剂量在给药后 2 小时可增加血浆花生四烯酸乙醇胺 (anandamide) 水平。在实验 2 中,ARN272 以 3.0 mg·mL−1(剂量为 9.0 mg·kg−1)的体积和 3.0 mg·mL−1(剂量为 18 mg·kg−1)的体积,分别输注给鼩鼱。选择该浓度是基于之前的实验,在这些实验中,S. murinus 所需的剂量至少是大鼠有效剂量的三倍。 在实验 1 中,SR141716 的浓度为 1.0 mg·mL−1(剂量为 1.0 mg·kg−1),输注体积为 1 mL·kg−1。选择 1.0 mg·kg−1 剂量的 SR141716 是基于其先前在逆转断点和恢复尼古丁自我给药方面的有效性,同时还发现该剂量不会增强催吐剂的作用,这与 2.5 mg·kg−1 或更高剂量的 SR141716 不同。 所有实验中使用的 0.15 M LiCl 药物处理均用无菌水配制,在实验 1 中,大鼠的给药体积为 20 mL·kg−1 (127 mg·kg−1),在实验 2 中,鼩鼱的给药体积为 60 mL·kg−1 (390 mg·kg−1)。 大鼠条件性张口实验方案:雄性 Sprague-Dawley 大鼠(单笼饲养,反转光暗周期)在异氟烷麻醉下进行口腔内插管手术植入。恢复后,大鼠接受2分钟的适应性水灌注。条件反射训练(两次训练,间隔72小时):大鼠在接受2分钟0.1%糖精溶液(1 mL/min)口内灌注前120分钟,预先腹腔注射ARN272(0.1、1.0或3.0 mg/kg)或溶剂(1:1:8 PEG400/Tween80/生理盐水),随后立即腹腔注射氯化锂(0.15 M,127 mg/kg)或生理盐水。另一些组在条件反射训练前30分钟腹腔注射SR141716(1.0 mg/kg)。第二次条件反射训练72小时后,进行无药物测试:大鼠接受2分钟糖精溶液灌注,并录像记录张口反应,评分者采用盲法。随后,对禁水大鼠进行单瓶条件性味觉厌恶(CTA)测试(分别在30分钟和120分钟时测量0.1%糖精的消耗量)。[1] 鼩鼱呕吐实验方案:家麝鼩(Suncus murinus,雌雄均有,体重26-53克)在注射氯化锂(LiCl,0.15 M,390 mg/kg,腹腔注射)前120分钟预先注射ARN272(9.0或18.0 mg/kg,腹腔注射)或溶剂。然后将鼩鼱放入有机玻璃箱中45分钟,由观察员现场计数呕吐次数(腹部收缩并排出胃液)。[1] |
| 参考文献 |
Br J Pharmacol.2013 Nov;170(5):1130-6.
|
| 其他信息 |
ARN272(CAS号:488793-85-7)是一种选择性花生四烯酸乙醇胺(anandamide)转运抑制剂,作用于FLAT(FAAH-1样花生四烯酸乙醇胺转运蛋白),在所用剂量下不会显著抑制FAAH活性。与也会影响其他N-酰基乙醇胺的FAAH抑制剂不同,ARN272提供了一种更具选择性的工具来研究花生四烯酸乙醇胺介导的作用。观察到的止吐和止吐作用依赖于CB₁受体。该化合物不干扰条件性味觉回避(CTA),证实其作用是选择性地抑制恶心,而非影响一般的联想学习。鼩鼱所需的剂量较高可能是由于其基础代谢率和肝脏清除率较高。[1]
|
| 分子式 |
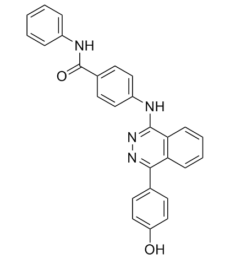
C27H20N4O2
|
|---|---|
| 分子量 |
432.473305702209
|
| 精确质量 |
432.158
|
| CAS号 |
488793-85-7
|
| PubChem CID |
1386351
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
629.1±55.0 °C at 760 mmHg
|
| 闪点 |
334.3±31.5 °C
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
| 折射率 |
1.757
|
| LogP |
4.58
|
| tPSA |
87.1
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
624
|
| 定义原子立体中心数目 |
0
|
| SMILES |
OC(C=C1)=CC=C1C2=NN=C(C3=C2C=CC=C3)NC4=CC=C(C(NC5=CC=CC=C5)=O)C=C4
|
| InChi Key |
UPKNGUQNXSMHBE-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C27H20N4O2/c32-22-16-12-18(13-17-22)25-23-8-4-5-9-24(23)26(31-30-25)28-21-14-10-19(11-15-21)27(33)29-20-6-2-1-3-7-20/h1-17,32H,(H,28,31)(H,29,33)
|
| 化学名 |
4-[[4-(4-hydroxyphenyl)phthalazin-1-yl]amino]-N-phenylbenzamide
|
| 别名 |
ARN272; ARN-272; ARN 272
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~125 mg/mL (~289.04 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.81 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.81 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3123 mL | 11.5615 mL | 23.1230 mL | |
| 5 mM | 0.4625 mL | 2.3123 mL | 4.6246 mL | |
| 10 mM | 0.2312 mL | 1.1561 mL | 2.3123 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Mean (+SEM) number of gapes by rats on drug-free test day, in experiment 1, by each of the groups.Br J Pharmacol.2013 Nov;170(5):1130-6. |
|---|
 Mean (±SEM) cumulative volume of saccharin consumed by rats 24 h after TR test day and immediately following water restriction, at 30 and 120 min, in experiment 1.Br J Pharmacol.2013 Nov;170(5):1130-6. |
 Mean (±SEM) number of vomiting episodes displayed byS. murinusduring the 45 min post-LiCl administration observation period.Br J Pharmacol.2013 Nov;170(5):1130-6. |
 GSK 525768A
GSK 525768A
 kb-NB77-78
kb-NB77-78
 GSK2807
GSK2807
 Eleclazine
Eleclazine
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
