| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
Forkhead box O1 (FoxO1) (IC50: 110 nM) [1]
|
|---|---|
| 体外研究 (In Vitro) |
在大鼠肝细胞系中,AS1842856 有效抑制人 Foxo1 反式激活,并通过抑制磷酸烯醇丙酮酸羧激酶和葡萄糖 6 磷酸酶 mRNA 的量来减少葡萄糖合成[1]。 AS1842856治疗后,p-Akt表达相对于对照组有所下降,但FoxO1和p-FoxO1的蛋白表达没有明显差异[2]。
AS1842856以剂量依赖方式抑制 FoxO1 依赖的转录活性。在转染 FoxO1 响应性荧光素酶报告质粒的 HEK293 细胞中,该化合物处理可显著降低荧光素酶活性,IC50 为 110 nM。此外,它可抑制肝细胞中 FoxO1 靶基因(如磷酸烯醇式丙酮酸羧激酶 PEPCK 和葡萄糖 - 6 - 磷酸酶 G6Pase)的表达 [1] 此外,我们进一步探讨了Akt、fox01和SIRT1之间的关系。我们分别用FoxO1抑制剂AS1842856 (0.2 μM)、Akt抑制剂MK-2206 (5 μM)或AS1842856联合MK-2206处理大鼠NP细胞。首先,MK-2206抑制了p-Akt的表达,降低了FoxO1在Ser256位点的磷酸化,进一步增加了FoxO1总蛋白的表达(图7A,B)。此外,AS1842856作为FoxO1的抑制剂,仅通过与FoxO1结合来降低FoxO1的活性,而不影响其转录[25]。然后在我们的实验中,AS1842856处理后,p-FoxO1和FoxO1的蛋白表达与对照组相比没有显著差异,但p-Akt的表达与对照组相比有所下降(图7A,B)。最后,单纯通过MK-2206抑制p-Akt活性可促进SIRT1的表达,而通过AS1842856抑制FoxO1活性可抑制SIRT1的表达。但如果同时抑制p-Akt和FoxO1,则SIRT1的表达受到抑制,这与单纯MK-2206处理组的结果不同(图7A,B)。[2] |
| 体内研究 (In Vivo) |
通过抑制肝糖异生基因,给糖尿病 db/db 小鼠口服 AS1842856 会导致空腹血糖水平显着降低;然而,给予正常小鼠的空腹血糖水平几乎没有影响。在正常小鼠和 db/db 小鼠中,AS1842856 治疗还可减少丙酮酸注射引起的血浆葡萄糖水平升高[1]。
在糖尿病 db/db 小鼠中,每日口服给予AS1842856(3、10、30 mg/kg),连续 14 天,可剂量依赖性改善空腹血糖。30 mg/kg 剂量组的空腹血糖水平较溶媒对照组显著降低。该化合物还有降低血浆胰岛素水平的趋势,但未达统计学显著性 [1] |
| 酶活实验 |
采用荧光素酶报告实验评估 FoxO1 抑制活性。HEK293 细胞共转染 FoxO1 表达质粒和含 FoxO1 结合元件的荧光素酶报告质粒。转染后,细胞用不同浓度的AS1842856处理 24 小时。随后检测荧光素酶活性,并将其归一化至蛋白质浓度,以确定对 FoxO1 依赖转录的抑制效果 [1]
|
| 细胞实验 |
分离原代肝细胞并进行培养。用不同浓度的AS1842856处理细胞特定时间后,提取总 RNA,通过实时 PCR 分析 FoxO1 靶基因(PEPCK 和 G6Pase)的 mRNA 表达水平。同时通过 Western blot 检测蛋白质水平,以验证对 FoxO1 介导信号通路的影响 [1]
|
| 动物实验 |
AS1842856溶于6%环糊精溶液中,用于口服给药。
糖尿病db/db小鼠 雄性db/db小鼠(8-10周龄)随机分组。AS1842856溶于0.5%甲基纤维素溶液中,每日一次口服给药,剂量分别为3、10或30 mg/kg,连续14天。对照组仅给予0.5%甲基纤维素溶液。在治疗前和治疗期间定期测量空腹血糖水平。研究结束时也测定了血浆胰岛素水平[1]。 |
| 参考文献 |
|
| 其他信息 |
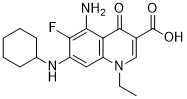
AS1842856 是一种喹诺酮类化合物,其结构为 4-喹诺酮,分别在 1、3、5、6 和 7 位被乙基、羧基、氨基、氟和环己基氨基取代。它能直接结合并阻断活性叉头框蛋白 O1 (Foxo1) 的转录活性,但不能结合 Ser256 磷酸化形式的 Foxo1。低浓度下,它能诱导伯基特淋巴瘤细胞系的细胞死亡和生长停滞。它具有降血糖、抗肥胖、自噬抑制、抗肿瘤、诱导细胞凋亡和抑制 Foxo1 等多种活性。它是一种喹啉单羧酸、喹诺酮、有机氟化合物、伯氨基化合物、仲氨基化合物和叔氨基化合物。
肝脏通过糖异生途径产生过多的葡萄糖是导致2型糖尿病(T2DM)患者血糖水平升高的部分原因。叉头转录因子叉头框蛋白O1(Foxo1)在介导胰岛素对肝脏糖异生的影响中起着至关重要的作用。本文利用db/db小鼠模型,证明了口服活性小分子化合物Foxo1抑制剂作为治疗T2DM药物的有效性。我们利用质谱亲和筛选法发现了一系列能与Foxo1结合的化合物,其中鉴定出一种化合物5-氨基-7-(环己基氨基)-1-乙基-6-氟-4-氧代-1,4-二氢喹啉-3-羧酸(AS1842856)。该化合物能有效抑制人Foxo1的转录激活,并通过抑制大鼠肝细胞系中葡萄糖-6-磷酸酶和磷酸烯醇式丙酮酸羧激酶的mRNA水平来降低葡萄糖生成。给糖尿病db/db小鼠口服AS1842856后,通过抑制肝脏糖异生基因,导致空腹血糖水平显著降低;而给正常小鼠服用AS1842856则对空腹血糖水平无影响。此外,AS1842856还能抑制正常小鼠和db/db小鼠注射丙酮酸后血糖水平的升高。综上所述,这些研究结果表明Foxo1抑制剂代表了一类可用于治疗2型糖尿病的新型药物。[1] 目的:氧化应激诱导的髓核(NP)细胞衰老是椎间盘退变(IDD)的重要原因之一。本研究旨在探讨沉默信息调节因子1(SIRT1)在氧化应激诱导的大鼠NP细胞衰老中的作用及其作用机制。方法:采用亚致死浓度的过氧化氢(H2O2)(100 μM)诱导大鼠NP细胞过早衰老。使用SRT1720(5 μM)激活SIRT1,以探讨其对NP细胞衰老的影响。分别使用AS1842856(0.2 μM)和MK-2206(5 μM)抑制FoxO1和Akt,以探讨Akt-FoxO1-SIRT1轴在大鼠NP细胞中的作用。本研究采用白藜芦醇(20 μM)预处理大鼠鼻咽部细胞,白藜芦醇是一种常见的抗氧化剂,也是SIRT1的间接激活剂,旨在探讨其在衰老大鼠鼻咽部细胞中的作用。结果显示,H2O2诱导的衰老大鼠鼻咽部细胞中SIRT1的mRNA和蛋白水平均降低,而SIRT1的特异性激活可抑制细胞衰老。Akt-FoxO1通路作为SIRT1的上游信号通路,可能通过影响SIRT1的表达参与H2O2诱导的大鼠鼻咽部细胞衰老的调控。此外,白藜芦醇在大鼠鼻咽部细胞中发挥抗衰老作用,这可能与Akt-FoxO1-SIRT1轴有关。结论:SIRT1通过Akt-FoxO1通路调控氧化应激诱导的大鼠鼻咽部细胞衰老,而白藜芦醇通过影响该信号通路发挥抗衰老作用。[2] |
| 分子式 |
C18H22FN3O3
|
|
|---|---|---|
| 分子量 |
347.383987903595
|
|
| 精确质量 |
347.165
|
|
| 元素分析 |
C, 62.23; H, 6.38; F, 5.47; N, 12.10; O, 13.82
|
|
| CAS号 |
836620-48-5
|
|
| 相关CAS号 |
|
|
| PubChem CID |
72193864
|
|
| 外观&性状 |
White to light yellow solid powder
|
|
| LogP |
3.839
|
|
| tPSA |
97.35
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
25
|
|
| 分子复杂度/Complexity |
562
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
O=C(C1C(=O)C2C(=CC(=C(C=2N)F)NC2CCCCC2)N(CC)C=1)O
|
|
| InChi Key |
MOMCHYGXXYBDCD-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C18H22FN3O3/c1-2-22-9-11(18(24)25)17(23)14-13(22)8-12(15(19)16(14)20)21-10-6-4-3-5-7-10/h8-10,21H,2-7,20H2,1H3,(H,24,25)
|
|
| 化学名 |
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.67 mg/mL (4.81 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 16.7 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 配方 2 中的溶解度: 1 mg/mL (2.88 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 10.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1 mg/mL (2.88 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 10 mg/mL (28.79 mM) in 6% HP-β-CD in Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8787 mL | 14.3935 mL | 28.7869 mL | |
| 5 mM | 0.5757 mL | 2.8787 mL | 5.7574 mL | |
| 10 mM | 0.2879 mL | 1.4393 mL | 2.8787 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
