| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
描述:BCI 盐酸盐 [(E)-BCI] 是一种新型、高效的 Dusp6(双特异性磷酸酶)变构抑制剂,它作用于磷酸酶结构域,阻止 ERK2 底物结合诱导的磷酸酶活性催化激活。在细胞中,它对 DUSP6 和 DUSP1 的 EC50 值分别为 13.3 μM 和 8.0 μM。
| 靶点 |
Dual specificity phosphatase 6 (DUSP6, also known as MKP3) and DUSP1 (MKP1) via allosteric inhibition. Inhibits ERK-stimulated DUSP6 activation. The compounds (including BCI and its analogs) inhibit DUSP6 and DUSP1 in the low micromolar range (IC₅₀ values in the micromolar range as shown in cellular complementation assays). Compound 7 (BCI-9) showed an EC₅₀ of 4.5 µM for FGF hyperactivation in zebrafish. Compound 19 (BCI-215) had an EC₅₀ of ~10 µM for FGF activation in zebrafish and inhibited DUSPs with IC₅₀ in the micromolar range. [1]
|
|---|---|
| 体外研究 (In Vitro) |
在 RAW264.7 巨噬细胞中,BCI 盐酸盐(100 ng/mL;24 小时)抑制 DUSP6 的表达 [2]。脂多糖 (LPS) 激活的巨噬细胞的 DUSP6 表达也受到 BCI 盐酸盐(0-1 nM;24 小时)的抑制。另一方面,BCI 盐酸盐(0-4 nM;24 小时)降低 ROS 的产生并刺激 Nrf2 的染色,从而激活 LPS 激活的巨噬细胞的吞噬作用 [2]。
在化学互补实验中,BCI 及其类似物(例如,7 (BCI-9)、19 (BCI-215))以浓度依赖的方式增加过表达 DUSP1 或 DUSP6 的 HeLa 细胞中 pERK 的水平,IC₅₀ 值在微摩尔范围内。 [1] 在利用重组DUSP6和ERK2进行的体外磷酸酶活性测定中,化合物BCI、7和19显著抑制了ERK刺激的DUSP6活化,但并未抑制基础磷酸酶活性。缺乏体内活性的醇类似物31(BCI-10)的抑制作用不显著。[1] 在利用EA.hy926细胞进行的细胞毒性测定中,浓度高于25 µM的BCI显示出细胞丢失、核浓缩和坏死(碘化丙啶染色)的迹象。相比之下,BCI-215在浓度高达50 µM时未表现出细胞毒性。[1] |
| 体内研究 (In Vivo) |
在dusp6启动子控制下表达GFP的转基因斑马鱼胚胎(Tg(dusp6:EGFP))中,BCI(20 µM)可过度激活FGF信号通路,通过自动图像分析(Cognition Network Technology)检测头部区域GFP荧光强度的增加来量化。处理5小时后观察到GFP表达达到最大值。[1] 在该斑马鱼模型中测试了一系列29种类似物。包括BCI在内的11种化合物表现出浓度依赖性的FGF信号通路过度激活作用。化合物7(BCI-9)活性最高(EC₅₀ 4.5 µM)。体内活性所必需的结构特征包括C-3位的脂肪族氨基烷基侧链和α,β-不饱和酮部分。 [1]化合物19 (BCI-215)在体内激活FGF信号通路(EC₅₀ ~10 µM),但即使在两倍EC₅₀浓度(20 µM)下,24小时暴露于斑马鱼胚胎后,也仅显示出极低的整体毒性。用化合物19处理的幼鱼正常孵化,而用EC₅₀浓度的BCI或化合物7处理的胚胎则未能孵化。[1]活性类似物在体内的FGF超激活活性与其在哺乳动物细胞实验中抑制DUSP6和DUSP1的能力相关。[1]
|
| 酶活实验 |
采用基于3-O-甲基荧光素磷酸酯(OMFP)的体外磷酸酶活性测定方法进行评估。将重组His标签DUSP6(250 ng)与化合物(100 µM)预孵育。为测定ERK2刺激的DUSP6活性,在加入OMFP(100 µM)启动反应前,向DUSP6/化合物混合物中加入重组ERK2(210 ng),最终反应体积为15 µL。在室温下,每隔10分钟测量一次荧光强度(激发/发射波长:485/525 nm),持续1小时。该测定方法评估化合物抑制底物(ERK2)诱导的DUSP6活化的能力。[1]
|
| 细胞实验 |
Western Blot 分析[2]
细胞类型: RAW264.7 巨噬细胞 测试浓度: 100 ng/mL 孵育时间: 24 小时 实验结果: 证实 DUSP6 蛋白下调。 RT-PCR[2] 细胞类型: RAW264.7 巨噬细胞 测试浓度: 0-1 nM 孵育时间: 24 小时 实验结果: 抑制 LPS 激活的巨噬细胞中 IL-1β 和 IL-6 mRNA 的表达。 使用基于哺乳动物细胞的化学互补试验评估 DUSP 的抑制作用。将 HeLa 细胞转染 Myc 标签的 DUSP1 或 DUSP6,并接种于 384 孔板中。48 小时后,将细胞以四复孔形式用化合物处理 15 分钟,然后用佛波酯 (TPA,500 ng/ml) 刺激 15 分钟以激活 ERK 通路。细胞用抗磷酸化ERK (pERK) 和c-Myc抗体进行免疫染色。使用荧光二抗可视化pERK和c-Myc-DUSP信号。通过多参数分析对孔板进行分析。根据c-Myc强度鉴定DUSP表达细胞。使用Kolmogorov-Smirnov (KS)统计量,通过比较处理孔和载体处理对照组的累积pERK分布来量化该亚群中的pERK水平。KS值越高,表明pERK恢复程度越高(即DUSP抑制程度越高)。生成剂量反应曲线,并计算IC₅₀值。[1] 在EA.hy926细胞中评估细胞毒性。将细胞接种于384孔板中,过夜使其贴壁,然后用化合物梯度处理6小时。随后,用碘化丙啶(PI,1 µg/ml)和Hoechst 33342(10 µg/ml)对细胞进行染色,分别标记坏死细胞和细胞核。进行活细胞成像,并测定细胞核计数、核浓缩程度和PI阳性细胞百分比等参数。[1] |
| 动物实验 |
斑马鱼(*Tg(dusp6:EGFP)*胚胎)用于体内构效关系(SAR)和毒性研究。胚胎通过自然交配获得,并在28.5°C下孵育。受精后24小时(hpf),将单个胚胎置于含有200 µL E3胚胎培养基的96孔板孔中。化合物以100倍浓度溶于DMSO中配制成储备液,并直接向孔中加入2 µL(最终DMSO浓度为1%)。在SAR研究中,胚胎用化合物处理,每个孔板均设置阴性对照(DMSO)。化合物处理后(通常为5小时,用于评估FGF激活),对胚胎进行麻醉,并使用配备4倍物镜(激发/发射波长:488/525 nm)的高内涵成像仪进行成像。使用自动图像分析软件对头部区域的GFP表达进行定量分析。为进行毒性评估,在初步成像后,将胚胎放回培养箱,并暴露于化合物中共24小时。随后对孔板进行目视检查,观察是否存在毒性迹象,例如明显的形态变化、坏死以及心跳或循环障碍。[1]
斑马鱼(Tg(dusp6:EGFP)胚胎)用于体内构效关系和毒性研究。胚胎通过自然交配获得,并在28.5°C下孵育。受精后24小时(hpf),将单个胚胎置于含有200 µL E3胚胎培养基的96孔板孔中。化合物以100倍浓度溶于DMSO中配制成储备液,并直接向孔中加入2 µL(最终DMSO浓度为1%)。在构效关系研究中,用化合物处理胚胎,并在每个孔板中设置阴性对照(DMSO)。化合物处理后(通常为 5 小时,用于评估 FGF 激活),对胚胎进行麻醉,并使用配备 4 倍物镜(激发/发射波长:488/525 nm)的高内涵读板仪进行成像。使用自动图像分析软件对头部区域的 GFP 表达进行定量。对于毒性评估,在初始成像后,将胚胎放回培养箱,并暴露于化合物中共 24 小时。然后对孔板进行目视检查,观察是否存在毒性迹象,例如明显的形态变化、坏死以及心跳或循环障碍。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
在斑马鱼胚胎中,许多活性类似物(包括BCI)在高剂量下表现出全身毒性,表现为明显的形态学改变、尾部弯曲表型以及暴露24小时后出现不透明的坏死细胞。吖啶橙染色证实了毒性,显示尾部存在死亡细胞。[1]
然而,毒性与α,β-不饱和酮部分的亲电性(通过Hammett σ常数测定)或体内靶点活性没有直接相关性。一些无活性类似物也具有毒性。[1] 化合物19(BCI-215)被鉴定为无毒类似物。其浓度为FGF激活EC₅₀(20 µM)的两倍时未显示毒性。与BCI或7处理的幼虫不同,用化合物19处理的幼虫在56 hpf时正常孵化。[1] 体外EA.hy926细胞毒性试验重现了这种差异性毒性:BCI在浓度高于25 µM时显示出毒性,而BCI-215在浓度高达50 µM时无毒性。[1] |
| 参考文献 |
|
| 其他信息 |
BCI是通过斑马鱼表型筛选发现的DUSP6变构抑制剂,凸显了斑马鱼在体内构效关系研究中的实用性。它通过与磷酸酶活性位点附近一个新型变构位点结合来抑制DUSP6,从而阻止ERK刺激激活所需的构象变化。分子建模表明,BCI的环己基氨基侧链和α,β-不饱和酮分别与DUSP6的Arg299和Trp264形成氢键。缺少酮基的醇类似物31无法形成这一关键氢键,因此没有活性。[1]该研究表明,可以通过变构机制靶向DUSP,从而克服其活性位点浅且保守以及对氧化还原敏感的催化半胱氨酸所带来的挑战。BCI及其类似物均能抑制DUSP6和DUSP1,表明这两种DUSP之间缺乏选择性。化合物 19 (BCI-215) 具有强效的 DUSP 抑制活性和极低的毒性,被认为是一种用于研究 DUSP1/DUSP6 生物学的改良化学探针。[1]
|
| 精确质量 |
352.147
|
|---|---|
| CAS号 |
95130-23-7
|
| 相关CAS号 |
BCI;1245792-51-1;(E/Z)-BCI;15982-84-0
|
| PubChem CID |
20831631
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 沸点 |
484.6ºC at 760mmHg
|
| 闪点 |
161.3ºC
|
| LogP |
2.324
|
| tPSA |
29.1
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
25
|
| 分子复杂度/Complexity |
470
|
| 定义原子立体中心数目 |
0
|
| SMILES |
Cl.O=C1C(=CC2C=CC=CC=2)C(NC2CCCCC2)C2C1=CC=CC=2
|
| InChi Key |
JPATUDRDKCLPTI-CRDKNBMZSA-N
|
| InChi Code |
InChI=1S/C22H23NO.ClH/c24-22-19-14-8-7-13-18(19)21(23-17-11-5-2-6-12-17)20(22)15-16-9-3-1-4-10-16/h1,3-4,7-10,13-15,17,21,23H,2,5-6,11-12H21H/b20-15-
|
| 化学名 |
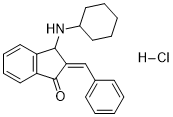
2-Benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one hydrochloride
|
| 别名 |
BCI hydrochloride
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~15.62 mg/mL (~44.14 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.56 mg/mL (4.41 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 15.6 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.56 mg/mL (4.41 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 15.6 mg/mL 澄清 DMSO 储备液加入 900 μL 20% SBE-β-CD 生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.56 mg/mL (4.41 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00507455 | COMPLETEDWITH RESULTS | Drug: solifenacin succinate Drug: tamsulosin hydrochloride Drug: Placebo to solifenacin Drug: Placebo to tamsulosin |
Bladder Outlet Obstruction Lower Urinary Tract Symptoms |
Astellas Pharma Inc | 2007-06 | Phase 2 |
| NCT00006034 | COMPLETED | Biological: keyhole limpet hemocyanin Drug: doxorubicin hydrochloride |
Bladder Cancer | Intracel | 1998-06 | Phase 3 |
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA

 463611831
463611831
