| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| Other Sizes |
|
| 靶点 |
PD-1/PD-L1 interaction
Human programmed death-ligand 1 (PD-L1) (blocks PD-1/PD-L1 interaction)[2] |
|---|---|
| 体外研究 (In Vitro) |
BMS-1001 将自身附着在人类 PD-L1 上并阻止其与 PD-1 相互作用。 BMS-1001 对所检查的细胞系表现出最小的毒性,并抑制可溶性 PD-L1 和细胞表面表达的 PD-1 之间的通讯。 BMS-1001 可减弱可溶性 PD-L1 对 T 细胞受体介导的 T 淋巴细胞活化的抑制作用 [1]。
BMS-1001 与人源PD-L1结合,并阻断其与PD-1的相互作用,这通过基于核磁共振的拮抗剂诱导解离实验证实。它能减轻可溶性及膜结合PD-L1对Jurkat T细胞(PD-1效应细胞)T细胞受体介导的激活的抑制作用。在使用PD-1效应细胞与表达PD-L1的人工抗原呈递细胞共培养的PD-1/PD-L1检查点实验中,BMS-1001 剂量依赖性地诱导荧光素酶活性,证明其在细胞界面拮抗PD-1/PD-L1检查点的能力,但其效力和最大激活水平低于治疗性抗体。[2] BMS-1001 剂量依赖性地消除了可溶性PD-L1 (sPD-L1) 对T细胞激活的抑制作用。在相对于sPD-L1为2倍和5倍摩尔过量的浓度下,它能将细胞激活完全恢复到仅使用抗CD3抗体观察到的水平。[2] 流式细胞术分析表明,将荧光标记的sPD-L1与BMS-1001 预孵育可显著降低其与表达PD-1的Jurkat细胞的结合,表明其在细胞表面干扰了PD-1/PD-L1相互作用。[2] BMS-1001 在溶液中诱导可溶性PD-L1形成二聚体,这通过NMR滴定(谱线增宽)、尺寸排阻色谱(保留时间偏移)和交联实验(SDS-PAGE显示二聚体条带)证实。[2] |
| 体内研究 (In Vivo) |
NP19 [[BMS-1001类似物]]在H22肝癌小鼠模型中的体内抗肿瘤活性[3]
鉴于NP19在黑色素瘤B16-F10肿瘤模型中具有良好的体内抗肿瘤效果,且PD-1/PD-L1抑制剂具有广谱的抗肿瘤活性,我们进一步利用H22肝癌模型BALB/c小鼠对复方NP19的体内抗肿瘤效果进行了评价。每只小鼠右侧皮下注射80万个H22细胞。当肿瘤体积达到约100 mm3时,随机选取小鼠,通过腹腔注射NP19或载体溶液治疗14天。如图8所示,NP19在25 mg/kg剂量下具有显著的体内抗肿瘤功效,TGI为76.5%(图8A, 8B, 8C)。此外,NP19没有引起明显的体重减轻(图8D),表明该化合物耐受性良好。 NP19 [[BMS-1001类似物]]在B16-F10小鼠黑色素瘤模型中的体内抗肿瘤活性[3] 为了确定新合成化合物的体外抗pd -1/PD-L1活性是否可以转化为体内功效,我们在小鼠黑色素瘤B16-F10肿瘤模型上测试了化合物NP19的抗肿瘤活性。选择NP19进行体内疗效研究,是因为与更有效的化合物NP2或同样有效的化合物NP12相比,NP19易于合成且细胞毒性较小(表9)。我们将携带黑色素瘤的BALB/c小鼠分别以载药对照和NP19 (25 mg/kg、50 mg/kg、100 mg/kg)灌胃治疗,每天1次,持续15天。如图6所示,治疗15天后,NP19治疗显著抑制了黑色素瘤的生长。 NP19的体内药代动力学性质[[BMS-10016的类似物]][3] 由于化合物NP19在体外表现出较高的药效,接下来通过静脉注射和口服给药来评估雄性Sprague-Dawley大鼠的药代动力学(PK)谱。表8总结了关键po和静脉给药PK参数。单次静脉给药1 mg/kg化合物NP19后,NP19的半衰期(t1/2)为1.5±0.5 h,清除率(CL)为0.9±0.2 L/h/kg,表观分布体积(Vss)为2.1±0.5 L/kg。NP19口服给药剂量为10 mg/kg时,观察到NP19的口服吸收(Tmax = 0.6±0.2 h)、长半衰期(t1/2 = 10.9±7.7 h)和口服生物利用度(F = 5%)。此外,在大鼠中未观察到明显的不良反应。NP19经口服灌胃后的半衰期(10.9 h)比静脉注射的半衰期(1.5 h)长得多;这可能是由于NP19的高亲脂性(logP = 7.9)或水溶性差。结果,NP19表现出翻转式药代动力学。这种翻转式的药代动力学有时会发生在水溶性较差的化合物中,如利巴米胺(43),由于水溶性较差(7.6 μg/mL),其t1/2 (p.o)/t1/2 (i.v)比为13.5。另一个例子是李建明等(44)报道的亲脂化合物IAT(一种水溶性为19 μg/mL的抗微管蛋白剂),其t1/2 (p.o.)/t1/2 (i.v.)的比值为~ 5,类似于NP19 [t1/2 (p.o.)/t1/2 (i.v.) = 7.1]。由于化合物NP19的口服生物利用度较低,我们推测需要高剂量才能提供足够的药物浓度以显示抗肿瘤功效。因此,我们进一步研究了化合物NP19的体内活性。 |
| 酶活实验 |
体外PD-1/PD-L1结合试验[3]
利用PD-1/PD-L1均质时间分辨荧光(HTRF)结合实验研究了化合物抑制PD-1/PD-L1相互作用的能力。PD-1/PD-L1结合检测试剂盒购自Cisbio。实验按照说明书进行,说明书可从https://www.cisbio.com/usa/drug-discovery/human-pd1pd-l1-biochemical-interaction-assay下载。 BMS 最近公开了第一个针对 PD-1/PD-L1 通路的非肽类小分子抑制剂,该抑制剂在均相时间分辨荧光 (HTRF) 结合测定中显示出活性;但没有提供支持其活动的进一步数据。 核磁共振测量[2] 在以15NH4Cl为唯一氮源的M9最小培养基中表达蛋白,获得均匀的15N标记。对于核磁共振测量,缓冲液通过凝胶过滤交换到pH为7.4 (PD-L1)的PBS或含有100 mM NaCl pH为6.4 (PD-1)的25 mM磷酸钠。在样品中加入10% (v/v)的D2O来提供锁定信号。所有光谱在300K下使用Bruker Avance III 600 MHz光谱仪记录。化合物与PD-L1的相互作用通过监测1h - 15n2d HMQC中核磁共振化学位移的扰动来评估。测试化合物解离PD-L1/PD-1的能力使用拮抗剂诱导解离试验进行评估。简而言之,15n标记的PD-1 (0.2 mM)与未标记的PD-L1稍微过滴定。这些化合物被合并到所得到的混合物中。实验过程中,h - 15n信号由HMQC监测。 PD1/PD-L1检查点试验[2] 检测前24小时,将aAPCs以每孔10 000个的密度在培养基中接种于96孔白色板中。在检测当天,在含有1% FBS的RPMI 1640中制备3.5倍连续稀释的抗体。在DMSO中制备了一系列BMS化合物[BMS-1001],并在含有1% FBS的RPMI 1640中配制。通过这种方法,所有样品中DMSO的浓度保持不变。从孔中取出95 μl的培养基,用40 μl的复合稀释液覆盖细胞。在含有1% FBS的40 μl RPMI 1640溶液中,每孔加入2万个ECs。37℃孵育6 h后,室温平衡10 min,每孔加入80 μl Bio-Glo试剂。孵育10分钟后,用FlexStation 3定量荧光。用Hill方程拟合实验数据,确定了一半最大有效浓度(EC50)和最大发光值(RLUmax)。 PD-1/ pd - l1效应试验[2] 为了评估BMS对可溶性PD-L1抑制T细胞的影响,在重组人PD-L1存在的情况下,用抗cd3抗体刺激ECs。为此,用5 μg/ml的抗cd3抗体或PBS中的同型对照液在96孔白色平底板上涂覆过夜,温度为4℃。除去抗体溶液,用PBS洗涤3次并干燥。将sPD-L1 (aa 18-134)在PBS中稀释,PBS中加入青霉素/链霉素溶液(每个溶液的终浓度为100 U/ml),存在BMS化合物[BMS-1001]或相应体积的DMSO。然后在抗体包被板的每孔中加入15 μl的溶液。ECs离心后稀释至5万/ ml,每孔加入细胞液60 μl。sPD-L1终浓度为10 μg/ml (0.6 μM)。BMS化合物[BMS-1001]的最终浓度分别为:0.12、0.3、1.2和3 μM,得到BMS:sPD-L1的摩尔比分别为1:5、1:2、2:1和5:1。细胞培养24小时,根据制造商的说明,使用Bio-Glo荧光素酶测定系统进行荧光素酶活性测定。 使用基于核磁共振的拮抗剂诱导解离实验评估BMS-1001破坏PD-1/PD-L1相互作用的能力。简而言之,将¹⁵N标记的PD-1与未标记的PD-L1轻微过滴定以形成复合物。向混合物中加入BMS-1001,并通过¹H-¹⁵N HMQC NMR监测。复合物的解离通过PD-1的尖锐¹H-¹⁵N信号的恢复来可视化,这些信号因复合物形成而变宽。[2] 使用直接NMR滴定评估BMS-1001与PD-L1的相互作用。用该化合物滴定均匀¹⁵N标记的PD-L1,监测¹H-¹⁵N HMQC谱中的化学位移变化和谱线增宽,以确认结合并表明化合物诱导的分子量增加(二聚化)。[2] |
| 细胞实验 |
细胞系[2]
为了验证BMS化合物[BMS-1001]抑制PD-1/PD-L1相互作用的效力,使用了基于细胞的PD-1/PD-L1免疫检查点阻断模型。在实验中,使用了两种模型细胞系:人工抗原呈递细胞(PD-L1+ aAPC/CHO-K1细胞,称为aAPCs)过表达TCR配体和PD-L1, T细胞替代品,一种经过修饰的过表达PD-1的Jurkat T细胞系,在NFAT启动子(PD-1效应细胞,称为ECs)的控制下携带荧光素酶报告细胞。从Promega中获得细胞,在添加10%胎牛血清、100 U/ml青霉素和100 U/ml链霉素的RPMI 1640培养基中培养。此外,细胞在持续存在的潮霉素B (50 μg/ml)和G418 (250 μg/ml)中繁殖,以提供引入的遗传构建物的稳定表达。后两种抗生素在实验中被省略。流式细胞术证实了ECs上PD-1和aAPCs上PD-L1的过表达(未示出),通过监测抗cd3抗体刺激后的荧光素酶活性证实了荧光素酶表达基因的存在。抗生素选择、流式细胞术和报告基因表达作为细胞系鉴定方法。使用基于pcr的方法对细胞进行周期性检测,发现支原体污染呈阴性。 细胞毒性试验[2] 将5 000个ECs接种在透明96孔板上,在BMS化合物[BMS-1001]浓度增加或DMSO作为对照(所有样品中DMSO浓度保持不变)的条件下培养48 h。处理后,根据制造商的说明,使用Biolog氧化还原染料混合MB进行代谢活性测试。 流式细胞术测定[2] 流式细胞术检测sPD-L1 (aa 18-134)与ECs的结合情况。将his标记的PD-L1蛋白或其突变体用NTA-Atto 647 N荧光染料在22°C下以8:1的摩尔比(蛋白:染料)染色2小时。将PD-L1-Atto与所测化合物或抗体在150 μl PBS中配制。样品在4°C黑暗中孵育30分钟。同时,将ECs离心,PBS洗涤,并以1 × 106个细胞/ml的浓度悬浮在新鲜PBS中,每个样品中加入50 μl ECs,冰孵育60 min,最终成分浓度为:PD-L1 (1.5 μM) 25 μg/ml,抗PD-L1抗体和对照抗体125 μg/ml, BMS化合物1 μM [BMS-1001]。使用BD FACS Verse流式细胞仪和BD FACSuite v1.0.6软件对样本进行分析。 细胞毒性实验: 将改造的Jurkat T细胞(PD-1效应细胞,ECs)接种于96孔板中,用递增浓度的BMS-1001或DMSO对照处理48小时。然后使用氧化还原染料混合物测量代谢活性,以确定细胞活力并计算EC₅₀值。BMS-1001显示出低毒性,EC₅₀为33.4 µM。[2] PD-1/PD-L1检查点实验: 将人工抗原呈递细胞(aAPCs,表达TCR配体和PD-L1的CHO-K1细胞)接种于白色96孔板中。将BMS-1001在DMSO中进行系列稀释,并用含1% FBS的RPMI培养基配制,保持DMSO浓度恒定。将PD-1效应细胞(ECs,表达PD-1和NFAT驱动的荧光素酶报告基因的Jurkat细胞)加入含有化合物稀释液的aAPCs中。在37°C共培养6小时后,使用荧光素酶检测试剂测量发光强度。发光表明从PD-1/PD-L1抑制中释放的TCR介导的激活。[2] PD-1/sPD-L1效应细胞实验: 将96孔板在4°C用抗CD3抗体包被过夜。将可溶性PD-L1 (sPD-L1) 与BMS-1001或DMSO在含有青霉素/链霉素的PBS中预孵育。将此混合物加入抗体包被的孔中。然后加入PD-1效应细胞(ECs)。培养24小时后,测量荧光素酶活性以评估T细胞激活。测试的BMS-1001最终浓度为0.12、0.3、1.2和3 µM,对应的BMS-1001与sPD-L1的摩尔比为1:5、1:2、2:1和5:1。[2] 流式细胞术结合实验: 将His标签的可溶性PD-L1用荧光NTA染料标记。将标记的PD-L1与BMS-1001、对照抗体或DMSO在PBS中预孵育。然后将表达PD-1的Jurkat细胞(ECs)加入混合物中并在冰上孵育。孵育后,通过流式细胞术分析细胞以测量荧光强度,指示sPD-L1与细胞表面PD-1的结合水平。[2] |
| 动物实验 |
雄性Sprague-Dawley大鼠药代动力学研究[3]
本研究采用雄性Sprague-Dawley大鼠(200-220 g)研究化合物NP19(BMS-1001类似物)的药代动力学。实验前12小时禁食,但可自由饮水。分别于口服(10 mg/kg)或静脉注射(1 mg/kg)化合物NP19后0.0833、0.25、0.5、1、1.5、2、4、6、8、12和24小时,从尾静脉采集血样(0.3 mL)至肝素化1.5 mL聚乙烯管中。该化合物溶于 5% DMSO 和 95% PEG-300 的混合溶液中用于静脉注射,或悬浮于 0.5% 羧甲基纤维素钠 (CMC-Na) 溶液中用于口服。样品立即以 3000 g 离心 10 分钟。所得血浆 (100 μL) 储存于 -20 °C 直至分析。使用 DAS(药物与统计)软件,通过非房室模型分析,根据个体动物数据确定药代动力学 (PK) 参数。PK 研究的仪器和分析条件:使用配备电喷雾电离 (ESI) 接口的 ACQUITY I-Class UPLC 和 XEVO TQD 三重四极杆质谱仪(Waters 公司,美国马萨诸塞州米尔福德)的 UPLC-MS/MS 系统分析血液样本。UPLC 系统由二元溶剂管理器 (BSM) 和带流通针的样品管理器 (SM-FTN) 组成。使用 Masslynx 4.1 软件(Waters 公司)进行数据采集和仪器控制。采用多反应监测 (MRM) 模式,NP19 的 m/z 555.35 → 181.03 和卡马西平的 m/z 237 → 194.1 进行定量分析。 小鼠 B16F10 黑色素瘤模型体内疗效研究[3] 使用 6-8 周龄的 BALB/c 小鼠研究 NP19(BMS-1001 的类似物)对皮下移植黑色素瘤细胞模型的抑制作用。将处于对数生长期的小鼠 B16F10 黑色素瘤细胞悬浮于 PBS 中,浓度为 2 × 10⁶ 个/mL。每只小鼠皮下接种 200 μL 含有 4 × 10⁵ 个细胞的细胞悬液。当肿瘤体积达到约100 mm³时,将小鼠随机分为四组(n = 10),分别给予NP19(25、50、100 mg/kg)和溶剂对照。药物通过灌胃法每日一次给药,持续15天。溶剂对照组灌胃0.5%羧甲基纤维素钠(CMC-Na)。在整个实验期间监测动物的活动和体重,以评估急性毒性。治疗开始16天后处死小鼠,收集肿瘤组织和主要器官(肝脏、脾脏、胸腺和肾脏)样本。收集的肿瘤组织和器官(肝脏、肾脏)用4%多聚甲醛固定,常规石蜡包埋,苏木精-伊红(H&E)染色,并在显微镜下观察。肿瘤生长抑制值 (TGI) 的计算公式为:TGI(%) = [1 – Wt/Wv] × 100%,其中 Wt 和 Wv 分别为治疗组和载体对照组的平均肿瘤重量。 小鼠 H22 肝癌模型体内疗效研究[3] 使用 6-8 周龄雄性 BALB/c 小鼠。根据肿瘤移植研究方案,将 8 × 10⁵ 个 H22 细胞接种到每只小鼠的右侧腹部。NP19(BMS-1001 的类似物)溶解于 5% DMSO、40% PEG-200 和 55% 生理盐水中,配制成所需浓度。对照组小鼠腹腔注射 200 μL 载体溶液。每隔2天使用可溯源的电子数显卡尺测量肿瘤体积,并使用公式a × b² × 0.5计算,其中a和b分别代表肿瘤的长径和短径。治疗结束后处死小鼠,切除肿瘤并称重。 |
| 毒性/毒理 (Toxicokinetics/TK) |
BMS-1001在基于细胞的试验中显示出较低的非特异性毒性。代谢活性测定结果显示,经48小时处理后,BMS-1001对修饰的Jurkat T细胞(PD-1效应细胞)的细胞毒性EC₅₀为33.4 µM。[2]
|
| 参考文献 |
|
| 其他信息 |
近年来,靶向PD-1/PD-L1免疫检查点的抗体在抗癌治疗中取得了显著成功。然而,目前尚未有具有细胞活性的小分子药物被报道。本文提供的证据表明,小分子药物能够缓解PD-1/PD-L1免疫检查点介导的Jurkat T淋巴细胞耗竭。由百时美施贵宝公司开发的两种优化的小分子PD-1/PD-L1相互作用抑制剂BMS-1001和BMS-1166,在分离的蛋白质上进行测试时,能够与人PD-L1结合并阻断其与PD-1的相互作用。这些化合物对所测试的细胞系毒性较低,并且能够阻断可溶性PD-L1与细胞表面表达的PD-1的相互作用。因此,BMS-1001 和 BMS-1166 可减轻可溶性 PD-L1 对 T 细胞受体介导的 T 淋巴细胞活化的抑制作用。此外,这些化合物还能有效减弱细胞表面相关 PD-L1 的抑制作用。我们还解析了 BMS-1001 和 BMS-1166 与 PD-L1 复合物的 X 射线晶体结构,揭示了这些化合物相比其前体化合物效力增强的可能原因。进一步的研究有望开发出基于口服免疫检查点抑制剂的抗癌疗法。[1]
使用单克隆抗体阻断 PD-1/PD-L1 免疫检查点通路已在癌症治疗领域取得了显著进展。然而,基于抗体的免疫疗法也存在一些缺点,例如抗体成本高昂、半衰期有限以及免疫原性。由于该通路结构信息不完整,开发能够克服这些缺陷的小分子PD-1/PD-L1抑制剂进展缓慢。百时美施贵宝公司近期公布了首批化学合成的PD-1/PD-L1抑制剂。本文介绍了这两类抑制剂的核磁共振和X射线晶体结构表征。PD-L1/抑制剂复合物的X射线晶体结构显示,一个抑制剂分子位于PD-L1同源二聚体的中心,填充在两个PD-L1分子之间一个深的疏水通道状口袋中。 (2-甲基-3-联苯基)甲醇衍生物的结构在通道的一侧被封堵,而基于[3-(2,3-二氢-1,4-苯并二恶英-6-基)-2-甲基苯基]甲醇的化合物则诱导扩大的相互作用界面,从而在PD-L1二聚体中形成开放的“面背式”隧道。[2] BMS-1001是由百时美施贵宝公司开发的一种PD-1/PD-L1免疫检查点小分子抑制剂。它属于一类旨在阻断PD-1和PD-L1相互作用从而恢复T细胞功能的化合物。[2] BMS-1001与人PD-L1的Ig样V型结构域复合物的晶体结构已在2.01 Å分辨率下解析。该化合物结合于PD-L1二聚体界面处的隧道中,诱导构象变化,包括Tyr56侧链的旋转。(2R)-2-氨基-3-羟基丙酸部分与Asp122形成氢键,而3-氰基苄基取代基与Tyr123和Arg125形成疏水相互作用。[2] BMS-1001诱导溶液中PD-L1的二聚化,这种机制被认为通过占据两个亚基上的PD-1结合位点而发挥其抑制作用。[2] 尽管已证实该化合物具有恢复T细胞活化的活性,但其效力低于治疗性抗PD-1/PD-L1抗体。[2] |
| 分子式 |
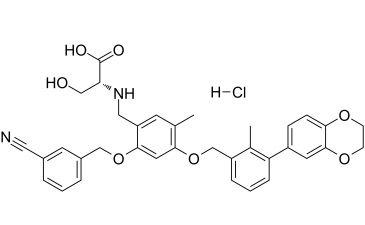
C35H35CLN2O7
|
|---|---|
| 分子量 |
631.1146
|
| 精确质量 |
630.21327
|
| 元素分析 |
C, 66.61; H, 5.59; Cl, 5.62; N, 4.44; O, 17.75
|
| CAS号 |
2113650-04-5
|
| 相关CAS号 |
2113650-03-4;2113650-04-5 (HCl);
|
| PubChem CID |
145925650
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
130Ų
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
12
|
| 重原子数目 |
45
|
| 分子复杂度/Complexity |
957
|
| 定义原子立体中心数目 |
1
|
| SMILES |
Cl[H].O(C([H])([H])C1C([H])=C([H])C([H])=C(C2C([H])=C([H])C3=C(C=2[H])OC([H])([H])C([H])([H])O3)C=1C([H])([H])[H])C1=C([H])C(=C(C([H])=C1C([H])([H])[H])C([H])([H])N([H])[C@@]([H])(C(=O)O[H])C([H])([H])O[H])OC([H])([H])C1C([H])=C([H])C([H])=C(C#N)C=1[H]
|
| InChi Key |
(2-((3-Cyanobenzyl)oxy)-4-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-methylbenzyl)oxy)-5-methylbenzyl)-D-serine hydrochloride
|
| InChi Code |
VKDUZONDVBDCAX-VNUFCWELSA-N SMILES
|
| 化学名 |
BMS-1001 HCl BMS 1001 HCl BMS1001 HCl BMS-1001 hydrochloride BMS 1001 hydrochloride BMS1001 hydrochloride
|
| 别名 |
BMS-1001 HCl; BMS 1001 HCl; BMS1001 HCl; 2113650-04-5; BMS-1001 hydrochloride; BMS-1001 (hydrochloride); BMS-1001 HCl; (2-((3-Cyanobenzyl)oxy)-4-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-methylbenzyl)oxy)-5-methylbenzyl)-D-serine hydrochloride; (2R)-2-[[2-[(3-cyanophenyl)methoxy]-4-[[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methoxy]-5-methylphenyl]methylamino]-3-hydroxypropanoic acid;hydrochloride; (2R)-2-[({2-[(3-cyanophenyl)methoxy]-4-{[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methoxy}-5-methylphenyl}methyl)amino]-3-hydroxypropanoic acid hydrochloride; N-[[2-[(3-cyanophenyl)methoxy]-4-[[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methoxy]-5-methylphenyl]methyl]-D-serine, monohydrochloride; BMS-1001 hydrochloride; BMS 1001 hydrochloride; BMS1001 hydrochloride
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~12.5 mg/mL (~19.81 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.25 mg/mL (1.98 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 12.5 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.25 mg/mL (1.98 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 12.5 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5845 mL | 7.9225 mL | 15.8451 mL | |
| 5 mM | 0.3169 mL | 1.5845 mL | 3.1690 mL | |
| 10 mM | 0.1585 mL | 0.7923 mL | 1.5845 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 PHPS1 Sodium
PHPS1 Sodium
 BE 2254
BE 2254
 环胞啶硫酸盐
环胞啶硫酸盐
 Azido-PEG5-acid
Azido-PEG5-acid
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
