| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
JAK1 (IC50 = 10 nM); JAK2 (IC50= 28 nM); Tyk2 (IC50= 116 nM); JAK3 (IC50= 810 nM)
Filgotinib (GLPG-0634) is a highly selective ATP-competitive inhibitor of Janus kinase 1 (JAK1), with minimal activity against JAK2, JAK3, and TYK2. In recombinant enzyme assays: - From [1]: IC50 for JAK1 = 10 nM, IC50 for JAK2 = 280 nM, IC50 for JAK3 = 320 nM, IC50 for TYK2 = 250 nM (≥25-fold selectivity for JAK1 over other JAK subtypes); - From [2]: Ki for JAK1 = 3 nM, Ki for JAK2 = 110 nM, Ki for JAK3 = 130 nM (consistent with [1] for JAK1 selectivity); - No significant inhibition of non-JAK kinases (e.g., EGFR, SRC, MAPK) at concentrations up to 1000 nM [1,2] |
|---|---|
| 体外研究 (In Vitro) |
filgotinib (GLPG0634) 剂量依赖性地抑制由 IL-4(一种通过 JAK1 和 JAK3 发出信号的细胞因子)介导的 Th2 细胞分化。此外,filgotinib 还在 1 μM 或更低的浓度下有效抑制 Th1 分化 [1]。 PRL 或 EPO 产生的 JAK2 同二聚体介导的信号传导 (IC50 > 10 μM) 不受 filgotinib (GLPG0634) 抑制 [2]。
T细胞JAK1-STAT信号抑制(来自[1]):在抗CD3/抗CD28刺激的人CD4+ T细胞中,Filgotinib(GLPG-0634) (1–100 nM)剂量依赖性抑制增殖:IC50 = 12 nM(72小时CFSE稀释法)。30 nM浓度下: - 降低磷酸化STAT3(p-STAT3,Tyr705)85%、磷酸化STAT1(p-STAT1,Tyr701)70%(蛋白质印迹法); - 酶联免疫吸附实验(ELISA)显示,促炎细胞因子IL-6减少65%、IFN-γ减少60%[1] - PBMC炎症反应抑制(来自[1]):在脂多糖(LPS,1 μg/mL)或IL-6(10 ng/mL)刺激的人外周血单个核细胞(PBMC)中,Filgotinib(GLPG-0634) (5–50 nM)抑制细胞因子驱动的信号: - 20 nM使LPS诱导的TNF-α减少55%、IL-1β减少50%; - 30 nM阻断IL-6诱导的p-STAT3(减少90%),并通过qPCR检测到急性期蛋白(CRP)mRNA减少65%[1] - JAK1酶学选择性(来自[2]):重组JAK家族酶实验中,Filgotinib(GLPG-0634) (0.1–1000 nM)对JAK1的选择性是JAK2/JAK3的>35倍,无脱靶激酶抑制(EGFR/SRC的IC50 > 1000 nM)[2] |
| 体内研究 (In Vivo) |
在经过修改的大鼠 CIA 模型中,filgotinib(GLPG0634;3、10、30 mg/kg,口服)剂量依赖性地抑制病程。 Filgotinib(50 mg/kg,op)抑制骨骼和软骨的恶化,有效减少足部T细胞(CD3+细胞)和巨噬细胞(F4/80+细胞)的浸润,并降低血液中细胞因子和趋化因子的水平,例如 IL-6、IP-10、XCL1 和 MCP-1[1]。在 CIA 大鼠模型中,filgotinib(GLPG0634;0.1 和 0.3 mg/kg)显示出有效性 [2]。
胶原诱导关节炎(CIA)小鼠疗效(来自[1]):DBA/1J CIA小鼠从免疫后21天开始给予Filgotinib(GLPG-0634) (10 mg/kg或30 mg/kg,口服,每日1次)处理: - 30 mg/kg使关节炎评分(0–16分制)从溶剂组8.3降至2.9(P<0.001); - 关节组织病理学显示,骨侵蚀减少70%、软骨丢失减少65%(较溶剂组); - 血清IL-6和TNF-α水平分别降低75%和60%[1] - 迟发型超敏反应(DTH)模型疗效(来自[1]):卵清蛋白(OVA)诱导DTH的BALB/c小鼠,给予Filgotinib(GLPG-0634) (5 mg/kg或20 mg/kg,口服,每日1次)处理7天: - 20 mg/kg使耳肿胀程度较溶剂组减少65%; - 耳组织匀浆中IFN-γ减少70%、IL-17减少65%[1] |
| 酶活实验 |
生化实验[1]
IC50测定。[1] 重组JAK1、TYK2、JAK2和JAK3 分别在50 mM HEPES (pH 7.5)、1 mM EGTA、10 mM MgCl2、2 mM DTT和0.01% Tween 20中进行活性测定。测定每组分中JAK蛋白的量,保持初始速度和随时间的线性。ATP浓度相当于实验Km值的4倍,底物浓度(光共轭的JAK-1(Tyr1023)肽)与实验测定的Km值对应。室温孵育90 min后,在Lance检测缓冲液中加入2 nM的euroium -anti-phosphotyrosine Ab 和10 mM的EDTA,测定磷酸化底物的量。在加入ATP之前,将酶与化合物在RT下预孵育60分钟,测定化合物的IC50值。 Kd的测定。[1] 解离常数在一家CRO公司测定。将具有快速解离率的荧光标记ATP模拟物(分别为JAK1、JAK2和JAK3的PRO13、PRO14和PRO13)与纯化的jak的JH1结构域一起在20 mM MOPS (pH 7.5)、1 mM DTT、0.01% Tween 20和500 mM hydroxyectoine(仅限JAK3)中孵育30分钟。将化合物(浓度范围为520 pM至1.1 μM)添加到100% DMSO中,并测量报告位移的时间依赖性。得到探针位移50%时对应的IC50值,并根据Cheng-Prusoff方程计算Kd值。 JAK激酶活性实验(基于HTRF,来自[1]): 1. 将纯化人JAK1/JAK2/JAK3/TYK2(各0.2 μg/mL)与生物素化STAT肽底物(JAK1/JAK2/TYK2用STAT3底物,JAK3用STAT5底物;各1 μg/mL)、ATP(10 μM)在实验缓冲液(50 mM Tris-HCl pH 7.5、10 mM MgCl₂、1 mM DTT)中37°C孵育15分钟。 2. 加入系列浓度的Filgotinib(GLPG-0634) (0.1–1000 nM),继续孵育30分钟。 3. 用20 mM EDTA终止反应,加入抗磷酸化STAT穴状化合物抗体和链霉亲和素-铕。 4. 检测时间分辨荧光(665 nm/620 nm比值),通过四参数逻辑回归计算IC50[1] - JAK1结合亲和力实验(基于SPR,来自[2]): 1. 通过胺偶联法将重组人JAK1激酶域固定在CM5传感器芯片上。 2. 将系列浓度的Filgotinib(GLPG-0634) (0.3–300 nM)溶于运行缓冲液(10 mM HEPES pH 7.4、150 mM NaCl、0.05% Tween-20),以30 μL/min流速注入芯片。 3. 记录传感图,使用BIAevaluation软件通过1:1结合模型计算解离常数(Ki)[2] |
| 细胞实验 |
细胞分析[1]
IL-4诱导STAT6磷酸化[1] 将THP-1细胞(ATCC TIB-202)与化合物在室温下预孵育1 h,与IL-4 (10 ng/ml)在室温下孵育60 min,并进行流式细胞术处理。细胞在Cytofix/Cytoperm缓冲液中固定,在Phosflow perm缓冲液III中冰透30分钟。阻断(Fc阻断试剂)后,用小鼠抗人pe标记的抗pSTAT6 Ab检测pSTAT6。 IL-2、IL-3和促红细胞生成素诱导STAT5磷酸化[1] NK-92细胞(ATCC CRL-2407) IL-2饥饿过夜,与化合物在37℃预孵育1小时,RT下IL-2 (1 ng/ml)刺激20分钟,并进行alphasgreen分析。将TF1细胞在含0.1% FBS的RPMI 1640中饥饿过夜,在室温下用化合物预孵育1小时,在室温下用IL-3 (30 ng/ml)刺激20分钟,并进行AlphaScreen分析。UT-7-红细胞生成素(EPO)细胞(UT-7的EPO依赖性衍生物;Centocor)与化合物在RT下预孵育1小时,用EPO (1 U/ml)刺激20分钟,然后进行alphasgreen分析。pSTAT5的测量基本上是根据制造商的协议使用AlphaScreen技术。 IFN-α和IFN-γ诱导STAT1磷酸化[1] STAT1 U2OS细胞(Invitrogen,目录号:K1469)与化合物在37℃下预孵育1 h,用30,000 U/ml IFN-αB2 (PBL IFN来源,目录号:11115-1)或20 ng/ml IFN-γ在37℃下裂解1小时(裂解缓冲液含有2 nM Tb-Ab),根据制造商的方案,在RT下孵育60分钟。pSTAT1通过时间分辨荧光共振能量转移检测。 催乳素诱导STAT5磷酸化[1] 22Rv1细胞(ATCC CW22Rv)饥饿过夜,用化合物预孵育,用催乳素(PRL)触发;500 ng/ml人PRL 20 min),用10 mM Tris-HCl (pH 7.5)、5 mM EDTA、150 mM NaCl、0.5% Triton X-100、50 mM NaF、30 mM焦磷酸钠、10%甘油缓冲液(含磷酸酶/蛋白酶抑制剂鸡尾酒)裂解,离心。细胞裂解液(180 μg)用于STAT5免疫沉淀(anti-STAT5 polyclonal Abs, C-17;蛋白A-Sepharose珠)。Western blotting后用密度分析法测定总STAT5和磷酸化STAT5。 IL-3/ jak2诱导Ba/F3细胞增殖[1] Ba/F3细胞(由V. Lacronique, Paris, France提供)依赖于IL-3和JAK2信号,与化合物在37℃孵育40 h,之后通过测量ATP含量来分析细胞增殖。 肿瘤抑制素m诱导的HeLa细胞STAT1报告基因检测[1] 用pSTAT1报告基因构建体转染HeLa细胞(ATCC CCL-2)。LR0127)。转染24 h后,用化合物孵育1 h,用抑癌素M (OSM)触发;33 ng / ml)。孵育20 h后,裂解细胞,根据供应商推荐使用荧光素酶SteadyLite试剂盒测定荧光素酶活性。同时,测定4 mg/ml 2-硝基苯β-d-半乳糖苷存在时β-半乳糖苷酶活性。 可拆卸的实验。[1] 从American Type Culture Collection中获得的HeLa和HCT116细胞用50 nM的ON-TARGETplus SMARTpool小干扰RNA (siRNA)转染人JAK1、JAK2、JAK3或TYK2,或用非靶向或gapdh阴性对照siRNA转染Invitrogen公司的Lipofectamine RNAiMAX转染试剂。转染4天后,将细胞饥饿过夜,用IL-6/sIL-6R(均为250 ng/ml)刺激20分钟,并根据制造商的方案使用alphasgreen技术检测pSTAT1水平。 T细胞分化的研究进展[1] 利用淋巴细胞密度梯度离心从健康供体的肉色被毛中分离PBMCs。使用初始CD4+ T细胞分离试剂盒II,通过消耗非T辅助细胞和记忆CD4+ T细胞进一步分离初始CD4+ T细胞。分离的初始CD4+ T细胞在细胞因子存在的情况下,用板结合的抗cd3 (3 μg/ml)和抗cd28 (5 μg/ml)抗体刺激其分化为Th1、Th2或Th17 Th亚群。在10 μg/ml抗il -4 Ab、10 ng/ml IL-2和10 ng/ml IL-12的作用下培养Th1细胞。在10 μg/ml抗ifn -γ Ab (Becton Dickinson)、25 ng/ml IL-4和10 ng/ml IL-2的作用下培养Th2细胞极化。对于Th17细胞极化,使用以下细胞因子的混合物:10 ng/ml IL-6, 10 ng/ml IL-1β, 1 ng/ml TGF-β和100 ng/ml IL-23。为了监测化合物对T细胞分化的影响,在T细胞分化开始时按指定浓度添加化合物。5 d后,使用RNeasy Mini试剂盒提取RNA,进行逆转录,并通过实时检测IFN-γ (Th1标记物)、IL-13 (Th2标记物)或IL-17F (Th17标记物)的表达来监测Th亚群分化程度。 CD4+ T细胞增殖实验(CFSE稀释法,来自[1]): 1. 从PBMC中分离人CD4+ T细胞,用CFSE(5 μM)37°C标记15分钟。 2. 标记T细胞(1×10⁵细胞/孔)接种于96孔板,用抗CD3(2 μg/mL)和抗CD28(1 μg/mL)刺激,同时加入Filgotinib(GLPG-0634) (1/5/10/30/100 nM)。 3. 72小时后,流式细胞术分析CFSE稀释程度评估增殖,计算IC50[1] - PBMC细胞因子ELISA实验(来自[1]): 1. 人PBMC(1×10⁶细胞/mL)接种于24孔板,用Filgotinib(GLPG-0634) (5/10/20/30/50 nM)预处理1小时。 2. 用LPS(1 μg/mL)或IL-6(10 ng/mL)刺激细胞,孵育24小时。 3. 收集培养上清,夹心ELISA法检测TNF-α/IL-6/IL-1β浓度[1] - p-STAT蛋白质印迹实验(来自[1]): 1. Jurkat T细胞(2×10⁵细胞/孔)用无血清培养基饥饿4小时,加入Filgotinib(GLPG-0634) (10/20/30 nM)处理1小时,再用IL-6(10 ng/mL)刺激30分钟。 2. 细胞用RIPA缓冲液裂解,30 μg蛋白经10% SDS-PAGE电泳后,用抗p-STAT3(Tyr705)和抗STAT3抗体孵育,ECL显色可视化[1] |
| 动物实验 |
大鼠每日剂量为 30 mg/kg;小鼠每日两次剂量为 50 mg/kg。在胶原诱导性关节炎 (CIA) 大鼠模型中,口服 GLPG0634 在 3 mg/kg 剂量下显示出对骨损伤的显著保护作用。从 1 mg/kg 开始,它显著减少了炎症细胞的浸润。药代动力学[1]
制剂[1] GLPG0634 的制剂为:静脉注射用聚乙二醇 200/0.9% NaCl (60/40; v/v) 溶液,口服用 0.5% (v/v) 甲基纤维素溶液,用于所有已描述的体内研究。化合物纯度经高效液相色谱法(HPLC)测定,>95%。动物。[1] 雄性Sprague Dawley大鼠(180–200 g)和CD1小鼠(23–25 g)分别购自Janvier和Harlan公司。给药前两天,大鼠在异氟烷麻醉下接受颈静脉置管手术。给药前至少禁食16小时,直至给药后4–6小时。给药前至少禁食12小时,直至给药后4小时。所有体内实验均在专用无特定病原体(SPF)设施(22°C)中进行。药代动力学研究[1] GLPG0634 的给药途径有两种:一种是经食管单次灌胃给药,剂量为 5 mg/kg(给药体积为 5 ml/kg);另一种是经尾静脉单次推注给药,剂量为 1 mg/kg(给药体积为 5 ml/kg)。在大鼠研究中,每组包含 3 只大鼠,并通过颈静脉采集血样。在小鼠研究中,每组包含 21 只小鼠(每个时间点 n = 3),在异氟烷麻醉下通过心脏穿刺采集血样。使用肝素锂作为抗凝剂,分别于 0.05、0.25、0.5、1、3、5 和 8 小时(静脉途径)以及 0.25、0.5、1、3、5、8 和 24 小时(口服)采集血液样本。 采用液相色谱-串联质谱法测定 GLPG0634 血浆浓度,定量下限为 2 ng/ml。采用WinNonlin软件进行非房室模型分析,计算药代动力学参数。 体内药理学[1] 啮齿类CIA模型[1] 动物[1] Dark Agouti大鼠(雌性,7-8周龄)和DBA/1J小鼠(雄性,6周龄)购自Janvier公司。 材料[1] CFA和IFA购自Difco公司(美国密歇根州底特律市)。使用牛II型胶原蛋白(CII)。所有其他试剂均为试剂级,所有溶剂均为分析纯。 CIA.[1] 实验开始前一天,用0.05 M乙酸配制CII溶液(2 mg/ml),并储存于4°C。免疫前,将等体积的IFA和CII在预冷的玻璃瓶中用均质器混合,玻璃瓶置于冰水浴中。对于大鼠CIA实验,在第1天和第8天分别于尾根部皮内注射0.2 ml乳剂。该免疫方法是在已发表方法的基础上改进而来。在疾病发作后(发作时平均临床评分2.5 ± 0.3;每组10只大鼠),每日口服GLPG0634,持续14天,以0.1–30 mg/kg的剂量范围测定其体内疗效。 TNF-α阻滞剂依那西普以10 mg/kg的剂量,每周三次腹腔注射给药。据报道,完全有效的剂量需要重复给药,剂量范围为3-9 mg/kg。在我们的Dark Agouti雌性大鼠模型中,每周三次腹腔注射10 mg/kg依那西普可使疾病达到正常化,这通过临床评分、炎症、骨吸收、滑膜炎和软骨损伤的评估得以证实。在第7天或第11天,通过眼眶后静脉丛取血200 μl,并在给药前以及给药后1、3和6小时(每个时间点n=2或3)采集血液样本,使用肝素锂作为抗凝剂,用于稳态药代动力学分析。处死动物后,取出后爪进行X射线分析和组织学检查。采用Tukey多重比较检验对GLPG0634的三项研究进行荟萃分析。每只大鼠的评分除以同一读数和研究中载体组的平均评分,再乘以 100。对于所有研究中接受相同剂量的所有动物,取每次读数的相对评分平均值。在小鼠 CIA 实验中,于第 1 天和第 21 天分别在尾根部皮内注射 0.2 ml IFA/CII 乳剂。该免疫方法是在已发表方法的基础上改进的。在疾病发作后(发病时平均临床评分 2.4 ± 0.6;每治疗组 10 只小鼠),每日口服 GLPG0634 14 天,以 50 mg/kg 的剂量范围,每日两次,测定 GLPG0634 的体内疗效。依那西普的给药以及药效学和药代动力学分析基本按照大鼠CIA模型所述方法进行。 CIA小鼠实验方案(引自[1]):1. DBA/1J小鼠(雄性,8-10周龄)于第0天皮下注射牛II型胶原蛋白(100 μg佐剂),第21天加强免疫。2. 第28天(关节炎发作:爪肿胀≥0.5 mm),将小鼠随机分为3组(每组n=6):- 赋形剂组:0.5%甲基纤维素PBS溶液,每日灌胃;- 菲戈替尼(GLPG-0634)10 mg/kg:溶于0.5%甲基纤维素,每日灌胃; - Filgotinib (GLPG-0634) 30 mg/kg:溶剂和给药途径与 10 mg/kg 组相同。 3. 治疗持续 21 天。每日测量关节炎评分和体重。安乐死时,取出关节进行组织病理学检查,并收集血清进行细胞因子 ELISA 检测 [1] - DTH 小鼠方案(引自 [1]): 1. BALB/c 小鼠(雌性,6-8 周龄)于第 0 天皮下注射 OVA(100 μg,佐剂)进行致敏。 2. 第 7 天,小鼠右耳皮内注射 OVA(50 μg,PBS 溶液)进行激发;左耳注射 PBS。 3. 从第0天到第7天,小鼠接受Filgotinib(GLPG-0634)(5 mg/kg或20 mg/kg,口服,每日一次)治疗。4. 第8天,用游标卡尺测量耳厚度;将耳组织匀浆后进行IFN-γ/IL-17 ELISA检测[1] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服后,菲戈替尼迅速吸收。菲戈替尼的血浆峰浓度中位数出现在给药后2-3小时,GS-829845的血浆峰浓度中位数出现在给药后5小时。菲戈替尼可在2-3天内达到稳态血药浓度,GS-829845可在4天内达到稳态血药浓度。食物似乎对菲戈替尼的吸收没有显著影响;因此,服用该药不受进食影响。重复口服200 mg菲戈替尼后,报告的菲戈替尼Cmax和AUCτ值分别为2.15 μg/mL和6.77 μg·h/mL。对于主要代谢物 GS-829845,报道的 Cmax 为 4.43 μg/mL,AUCτ 为 83.2 μg·h/mL。 在总给药剂量中,约 87% 经肾脏排泄,15% 经粪便排泄。 代谢/代谢物 羧酸酯酶参与 filgotinib 的代谢。羧酸酯酶 2 (CES2) 同工酶主要负责将 filgotinib 代谢为其主要代谢物 GS-829845。虽然羧酸酯酶 1 (CES1) 在 filgotinib 的生物转化中作用较小,但体外研究表明,当 CES2 饱和时,CES1 可以部分补偿。 GS-829845 是目前唯一被鉴定的主要循环代谢物。 生物半衰期 filgotinib 的半衰期估计为 7 小时,而其活性代谢物 GS-829845 的半衰期估计为 19 小时。 大鼠口服生物利用度(引自 [1]):雄性 Sprague-Dawley 大鼠(250–300 g)通过灌胃(10 mg/kg)或静脉注射(2 mg/kg)接受 Filgotinib (GLPG-0634):- 口服生物利用度 = 62%;- 口服给药:Cmax = 3.8 μg/mL(Tmax = 1.5 h),末端半衰期 (t1/2) = 4.2 h,AUC0-24h = 20.7 μg·h/mL; - 静脉给药:Cmax = 9.5 μg/mL,t1/2 = 3.9 h,AUC0-∞ = 33.4 μg·h/mL [1] - 血浆蛋白结合率(引自[1]):在人血浆中,Filgotinib (GLPG-0634) 的蛋白结合率为 92%(通过 37°C 平衡透析法测定)[1] - CIA 小鼠体内的组织分布(引自[1]):CIA 小鼠口服 Filgotinib (GLPG-0634) (30 mg/kg) 后 2 小时,关节组织浓度为 4.5 μg/g,脾脏浓度为 4.2 μg/g,约为血浆浓度 (3.7 μg/mL) 的 1.2 倍 [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
妊娠期和哺乳期影响
◉ 哺乳期用药概述 菲戈替尼尚未获得美国食品药品监督管理局 (FDA) 的批准。目前尚无关于菲戈替尼在哺乳期临床应用的信息。欧洲制造商建议在接受filgotinib治疗期间停止母乳喂养。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白质结合 filgotinib约有55-59%与蛋白质结合,而其活性代谢物GS-829845的蛋白质结合率为39-44%。 啮齿动物重复给药毒性(来自[1]):雄性/雌性Sprague-Dawley大鼠(每性别每组n=4)接受Filgotinib(GLPG-0634)(5/30/100 mg/kg,口服,每日一次)治疗28天:- 无死亡;未观察到不良反应剂量 (NOAEL) = 30 mg/kg;- 100 mg/kg 时:轻度淋巴细胞减少症(淋巴细胞计数较对照组减少 20%),肝脏/肾脏无组织病理学变化;血清 ALT/AST/肌酐水平不变 [1] - 炎症模型中的体内安全性(来自 [1]):在 CIA 和 DTH 小鼠中(口服剂量高达 30 mg/kg,持续 21 天):- 无明显体重减轻(<4%);- 无明显毒性(例如,嗜睡、腹泻); - 血清肌酐和尿素氮(肾功能)保持正常[1] - 体外正常细胞安全性(引自[1]):用 Filgotinib (GLPG-0634) (≤100 nM) 处理 72 小时的人真皮成纤维细胞和外周血单核细胞 (PBMC) 显示出 >90% 的细胞活力(MTT 法)[1] |
| 参考文献 | |
| 其他信息 |
药效学
除了靶向抑制 Janus 激酶 (JAK) 1 外,filgotinib 还通过抑制 IL-6 诱导的 STAT1 磷酸化来靶向促炎细胞因子信号通路。filgotinib 给药后,血清 C 反应蛋白水平也会降低。 作用机制(引自 [1,2]):Filgotinib (GLPG-0634) 通过与 ATP 竞争激酶结构域来选择性抑制 JAK1,从而阻断 JAK1 介导的 STAT (STAT1/STAT3) 磷酸化。这可以抑制促炎细胞因子信号传导(IL-6/IFN-γ)和T细胞活化,从而减轻自身免疫性疾病的炎症[1,2] - 药物化学背景(引自[2]):Filgotinib (GLPG-0634) 是一种三唑并吡啶衍生物,由先导化合物优化而来,旨在增强其对JAK1的选择性(通过吡啶环的结构修饰)并提高口服生物利用度(降低首过代谢)[2] - 治疗潜力(引自[1]):临床前数据支持Filgotinib (GLPG-0634) 用于治疗JAK1驱动的炎症性疾病,包括类风湿性关节炎 (RA) 和银屑病。其高JAK1选择性可最大限度地减少脱靶效应(例如,JAK2介导的骨髓抑制)[1] |
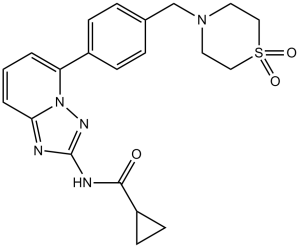
| 分子式 |
C21H23N5O3S
|
|---|---|
| 分子量 |
425.50
|
| 精确质量 |
425.152
|
| 元素分析 |
C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
|
| CAS号 |
1206161-97-8
|
| 相关CAS号 |
GLPG0634 analog;1206101-20-3;Filgotinib maleate;1802998-75-9;Filgotinib-d4;2041095-50-3; 1206161-97-8; 1540859-07-1 (HCl hydrate)
|
| PubChem CID |
49831257
|
| 外观&性状 |
Off-white to gray solid powder
|
| 密度 |
1.5±0.1 g/cm3
|
| 折射率 |
1.748
|
| LogP |
0.79
|
| tPSA |
108.28
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
715
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C1CC1C(=O)NC2=NN3C(=N2)C=CC=C3C4=CC=C(C=C4)CN5CCS(=O)(=O)CC5
|
| InChi Key |
RIJLVEAXPNLDTC-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H23N5O3S/c27-20(17-8-9-17)23-21-22-19-3-1-2-18(26(19)24-21)16-6-4-15(5-7-16)14-25-10-12-30(28,29)13-11-25/h1-7,17H,8-14H2,(H,23,24,27)
|
| 化学名 |
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide.
|
| 别名 |
GLPG-0634; PubChemSID 163643231; GLPG0634; 1206101-20-3; Filgotinib; GLPG0634; 1206161-97-8; N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide; Filgotinib (GLPG0634); N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide; GLPG 0634; Filgotinib
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.88 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.88 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.88 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.5 mg/mL (5.88 mM) (饱和度未知) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 5 中的溶解度: ≥ 2.5 mg/mL (5.88 mM) (饱和度未知) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 6 中的溶解度: 4% DMSO+30% PEG 300+ddH2O: 3mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3502 mL | 11.7509 mL | 23.5018 mL | |
| 5 mM | 0.4700 mL | 2.3502 mL | 4.7004 mL | |
| 10 mM | 0.2350 mL | 1.1751 mL | 2.3502 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link:https://clinicaltrials.gov/ct2/show/NCT07554495
Conditions:Polyarticular Course Juvenile Idiopathic Arthritishttps://clinicaltrials.gov/ct2/show/NCT07553182
https://clinicaltrials.gov/ct2/show/NCT06865417
Title:Prospective Observational Study of Effectiveness and Safety of Filgotinib in Participants With Ulcerative Colitis (UC)
Status:Active, not recruiting
UpdateDate:2025-12-24
Ctid:NCT05817942
Link:
https://clinicaltrials.gov/ct2/show/NCT05817942
https://clinicaltrials.gov/ct2/show/NCT05323591
https://clinicaltrials.gov/ct2/show/NCT06222034
https://clinicaltrials.gov/ct2/show/NCT05785611
https://clinicaltrials.gov/ct2/show/NCT04871919
https://clinicaltrials.gov/ct2/show/NCT04985435
https://clinicaltrials.gov/ct2/show/NCT03025308
https://clinicaltrials.gov/ct2/show/NCT02914535
https://clinicaltrials.gov/ct2/show/NCT06687551
https://clinicaltrials.gov/ct2/show/NCT03201445
https://clinicaltrials.gov/ct2/show/NCT05479058
https://clinicaltrials.gov/ct2/show/NCT06527534
https://clinicaltrials.gov/ct2/show/NCT02914600
https://clinicaltrials.gov/ct2/show/NCT06285539
https://clinicaltrials.gov/ct2/show/NCT02065700
https://clinicaltrials.gov/ct2/show/NCT03926195
https://clinicaltrials.gov/ct2/show/NCT02914561
https://clinicaltrials.gov/ct2/show/NCT06043739
https://clinicaltrials.gov/ct2/show/NCT05502731
https://clinicaltrials.gov/ct2/show/NCT04608344
https://clinicaltrials.gov/ct2/show/NCT04115748
https://clinicaltrials.gov/ct2/show/NCT03320876
https://clinicaltrials.gov/ct2/show/NCT03077412
https://clinicaltrials.gov/ct2/show/NCT04115839
https://clinicaltrials.gov/ct2/show/NCT03207815
https://clinicaltrials.gov/ct2/show/NCT05090410
https://clinicaltrials.gov/ct2/show/NCT03046056
https://clinicaltrials.gov/ct2/show/NCT02889796
https://clinicaltrials.gov/ct2/show/NCT02886728
https://clinicaltrials.gov/ct2/show/NCT02873936
https://clinicaltrials.gov/ct2/show/NCT02914522
https://clinicaltrials.gov/ct2/show/NCT03417778
https://clinicaltrials.gov/ct2/show/NCT04483687
https://clinicaltrials.gov/ct2/show/NCT04483700
https://clinicaltrials.gov/ct2/show/NCT01894516
https://clinicaltrials.gov/ct2/show/NCT01888874
https://clinicaltrials.gov/ct2/show/NCT03100942
https://clinicaltrials.gov/ct2/show/NCT03134222
https://clinicaltrials.gov/ct2/show/NCT03285711
https://clinicaltrials.gov/ct2/show/NCT02885181
https://clinicaltrials.gov/ct2/show/NCT03117270
https://clinicaltrials.gov/ct2/show/NCT03101670
https://clinicaltrials.gov/ct2/show/NCT02048618
https://clinicaltrials.gov/ct2/show/NCT02162355
https://clinicaltrials.gov/ct2/show/NCT02084199
https://clinicaltrials.gov/ct2/show/NCT01668641
https://clinicaltrials.gov/ct2/show/NCT01798979
https://clinicaltrials.gov/ct2/show/NCT01384422
https://clinicaltrials.gov/ct2/show/NCT01419990
https://clinicaltrials.gov/ct2/show/NCT01179581
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2018-003933-14
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2017-000402-38
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2017-000545-52
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2017-001485-17
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-003153-15
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-003558-34
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-003630-25
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-003179-23
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-002763-34
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-001367-36
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-000569-21
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-001392-78
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-002765-58
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-003637-14
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-003636-21
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-001496-75
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-000570-37
https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-000568-41
GLPG0634 inhibits the differentiation of Th1, Th2, and Th17 cells.J Immunol.2013 Oct 1;191(7):3568-77. |
GLPG0634 dose-dependently prevents disease progression in the therapeutic rat CIA model.J Immunol.2013 Oct 1;191(7):3568-77. |
GLPG0634 is efficacious in a mouse therapeutic CIA model.J Immunol.2013 Oct 1;191(7):3568-77. |
 JAK2-IN-14
JAK2-IN-14
 JAK1/TYK2-IN-5
JAK1/TYK2-IN-5
 Gusacitinib-d4
Gusacitinib-d4
 Povorcitinib-d6
Povorcitinib-d6
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
")
")
")
 463611831
463611831
