| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
CDK1 (IC50 = 40 nM); CDK2 (IC50 = 40 nM); CDK4 (IC50 = 40 nM); CDK6 (IC50 = 40 nM); CDK7 (IC50 = 300 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:最初发现 Flavopiridol 可抑制表皮生长因子受体和蛋白激酶 A(IC50 = 21 和 122 μM)。随后,当 Flavopiridol 在国家癌症研究所开发治疗计划的 60 个人类肿瘤细胞系小组中进行测试时,发现 Flavopiridol 在更生理相关的浓度 (IC50 = 66 nM) 下可抑制细胞增殖。 Flavopiridol 以时间和浓度依赖性方式抑制人乳腺癌细胞中的 CDK2 和 CDK4,从而诱导 G1 期阻滞。 Flavopiridol 的短时间治疗(约 12 小时)可诱导造血细胞系凋亡,包括 SUDHL4、SUDHL6(B 细胞系)、Jurkat 和 MOLT4(T 细胞系)以及 HL60(骨髓细胞)。在克隆形成试验中,Flavopiridol 是一种高效细胞毒性化合物,在 23 个人类肿瘤模型中的平均 IC70 为 8 ng/mL。最近的一项研究表明 Flavopiridol 治疗可诱导人胶质母细胞瘤 T98G 细胞系中大量 AKT-Ser473 磷酸化。激酶测定:如下在微量滴定板中测定CDK活性。将 40 μg Gst-Rb 与不同量的 Flavopiridol 和未标记的 ATP 混合。然后通过添加从表达重组人 CDK 的昆虫细胞获得的 S100 级分的硫酸铵片段来启动反应。最终反应条件为 10 mM MgCl2、50 mM Tris-HCl (pH 7.5) 和 1 mM DTT。 ATP 的终浓度相应调整。放射性标记的 ATP 用作磷酰基供体。添加酶后,反应在30°C下进行2.5分钟,然后添加EDTA终止反应。然后用谷胱甘肽-琼脂糖捕获 Gst-Rb,并通过液体闪烁计数测定掺入的放射性。细胞测定:以 1 × 106 个细胞/mL 的密度生长的细胞暴露于不同浓度和时间段的 Flavopiridol。提取DNA。简而言之,用冷磷酸盐缓冲盐水 (PBS) 洗涤细胞一次,并用 3 mL 裂解缓冲液(5 mM Tris-HCL [pH 7.5]; 20 mM EDTA; 0.5% Triton X-100)在 4° 裂解 15 分钟C。通过离心(26,000g、4°C 20 分钟)分离细胞裂解物的染色质。含有小 DNA 片段的上清液依次用苯酚、苯酚:氯仿 (1:1) 和氯仿提取。核酸在 0.5 M 氯化钠、90% 乙醇中于 -20 °C 过夜沉淀。然后用牛 RNAaseA (60 μg/mL) 消化 RNA。依次进行再提取和再沉淀后,将 DNA 溶解在 10 mM Tris-HCL (pH 7.5)、1 mM EDTA、0.5% 十二烷基硫酸钠 (SDS) 中,然后在 1.6% 琼脂糖凝胶上进行电泳。
|
| 体内研究 (In Vivo) |
在第 1-4 天和第 7-11 天口服最大耐受剂量 10 mg/kg/天时,Flavopiridol 可使 PRXF1337 中的肿瘤消退,并使 PRXF1369 中的肿瘤停滞持续 4 周。连续 5 天每天静脉内 (IV) 或腹膜内注射 7.5 mg/kg Flavopiridol 治疗后,12 个晚期皮下 (sc) 人 HL-60 异种移植物中的 11 个经历完全消退,几个月后动物仍保持无病状态。黄酮吡醇治疗一个疗程。用 7.5 mg/kg 静脉推注黄吡醇治疗 SUDHL-4 sc 淋巴瘤 5 天,经历主要(八只小鼠中的两只)或完全(八只小鼠中的四只)消退,其中两只动物在超过 60 年内保持无病状态。天。总体增长延迟为73.2%。每日静脉注射或腹腔注射黄吡醇可使血浆浓度峰值达到约 7 µM,随后在 8 小时内逐渐下降至约 100 nM。
|
| 酶活实验 |
以下是如何在微量滴定板中测量 CDK 活性。制备 40 μg Gst-Rb、不同浓度的 Flavopiridol 和未标记 ATP 的组合。随后,添加从表达人 CDK 重组体的昆虫细胞获得的 S100 级分的硫酸铵片段以引发反应。使用 10 mM MgCl2、50 mM Tris-HCl (pH 7.5) 和 1 mM DTT 的最终混合物。结果,最终 ATP 浓度发生改变。使用磷酰基供体,它是放射性标记的 ATP。添加酶后,反应在 30 °C 下运行 2.5 分钟,然后添加 EDTA 停止反应。用谷胱甘肽琼脂糖捕获 Gst-Rb 后,使用液体闪烁计数来计算已掺入的放射性。
|
| 细胞实验 |
对以 1 × 106 细胞/mL 密度生长的细胞给予不同浓度和暴露时间的黄吡醇。它提取DNA。简而言之,细胞在 3 mL 裂解缓冲液(5 mM Tris-HCL [pH 7.5];20 mM EDTA;0.5% Triton X-100)中于 4 °C 裂解 15 分钟。此过程涉及使用磷酸盐缓冲盐水 (PBS) 进行一次冷洗。离心用于从细胞裂解物中分离染色质(26,000g、4°C 20 分钟)。使用以下方法提取含有小 DNA 片段的上清液:苯酚、苯酚:氯仿 (1:1) 和氯仿。 -20°C 过夜,核酸在 0.5 M 氯化钠和 90% 乙醇中沉淀。之后,牛 RNAaseA (60 μg/mL) 会分解 RNA。将 DNA 溶解在 10 mM Tris-HCL (pH 7.5)、1 mM EDTA 和 0.5% 十二烷基硫酸钠 (SDS) 中,然后连续进行再提取和再沉淀,然后在 1.6% 琼脂糖凝胶上进行电泳。
|
| 动物实验 |
人前列腺癌异种移植瘤PRXFI337和PRXFI369,皮下移植于裸鼠体内[4];人早幼粒细胞白血病HL-60、人B细胞滤泡淋巴瘤SUDHL-4和获得性免疫缺陷综合征(AIDS)-r
10 mg/kg/d;7.5 mg/kg/d 口服[4];腹腔注射或静脉注射 |
| 药代性质 (ADME/PK) |
代谢/代谢物
黄酮吡啶醇已知的人体代谢物包括(2S,3S,4S,5R)-6-[2-(2-氯苯基)-5-羟基-8-[(3S,4R)-3-羟基-1-甲基哌啶-4-基]-4-氧代色烯-7-基]氧基-3,4,5-三羟基氧杂环己烷-2-羧酸。 口服生物利用度:人体约20%;大鼠口服25 mg/kg后约30%[1][5] -消除半衰期:人体6-8小时;大鼠4.5小时(腹腔注射);小鼠体内半衰期为7.2小时(口服给药)[1][5] - 血浆蛋白结合率:在人血浆中>99%(浓度范围:0.1-10 μg/mL)[1][9] - 分布:大鼠体内分布容积 (Vd) = 3.5 L/kg,广泛分布于肿瘤组织、骨髓和淋巴器官[5][9] - 代谢:主要在肝脏中经CYP3A4代谢为无活性代谢物(例如,单羟基化衍生物)[1][5] - 排泄:70%的剂量以代谢物形式经粪便排出;25%经尿液排出;<2%以原形排出[1][5] |
| 毒性/毒理 (Toxicokinetics/TK) |
急性毒性:大鼠口服LD50 > 200 mg/kg;小鼠 > 150 mg/kg。- 腹腔注射LD50 = 大鼠 50 mg/kg;小鼠 40 mg/kg。- 亚慢性毒性(大鼠28天口服给药):剂量高达25 mg/kg/天时未见明显的肝毒性或肾毒性;50 mg/kg/天时出现轻度中性粒细胞减少症(中性粒细胞计数减少≤15%)和血小板减少症(血小板计数减少≤10%)。- 在异种移植小鼠中,治疗剂量(10-25 mg/kg/天)未引起明显的体重减轻或行为异常。- 药物相互作用:强效CYP3A4抑制剂(例如酮康唑)可抑制本品,使AUC增加2.2倍。与高剂量化疗药物(例如多柔比星)产生协同毒性
|
| 参考文献 |
|
| 其他信息 |
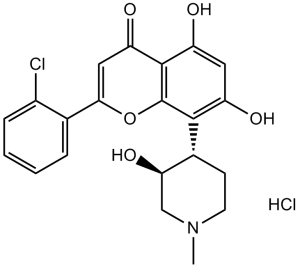
盐酸阿沃西地是等摩尔量的阿沃西地与氯化氢反应生成的盐酸盐。它是一种细胞周期蛋白依赖性激酶9 (CDK9) 抑制剂,已被研究用于治疗急性髓系白血病、关节炎和动脉粥样硬化斑块形成。它具有抗肿瘤、抗风湿、诱导细胞凋亡和抑制细胞周期蛋白依赖性激酶 (EC 2.7.11.22) 的作用。它含有阿沃西地(1+)离子。
盐酸阿沃西地是一种合成的N-甲基哌啶基氯苯基黄酮化合物。作为细胞周期蛋白依赖性激酶抑制剂,阿沃西地通过阻止细胞周期蛋白依赖性激酶(CDK)的磷酸化并下调细胞周期蛋白D1和D3的表达,诱导细胞周期停滞,最终导致G1期细胞周期阻滞和细胞凋亡。该药物也是三磷酸腺苷(ATP)活性的竞争性抑制剂。 |
| 分子式 |
C21H20CLNO5.HCL
|
|
|---|---|---|
| 分子量 |
438.3
|
|
| 精确质量 |
437.079
|
|
| 元素分析 |
C, 57.55; H, 4.83; Cl, 16.18; N, 3.20; O, 18.25
|
|
| CAS号 |
131740-09-5
|
|
| 相关CAS号 |
131740-09-5 (HCl);146426-40-6;
|
|
| PubChem CID |
9910986
|
|
| 外观&性状 |
Light yellow to khaki solid powder
|
|
| 沸点 |
603.6ºC at 760 mmHg
|
|
| 熔点 |
169.5-170ºC
|
|
| 闪点 |
318.8ºC
|
|
| 蒸汽压 |
2.03E-15mmHg at 25°C
|
|
| LogP |
4.044
|
|
| tPSA |
94.14
|
|
| 氢键供体(HBD)数目 |
4
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
29
|
|
| 分子复杂度/Complexity |
628
|
|
| 定义原子立体中心数目 |
2
|
|
| SMILES |
ClC1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C(C2C(=C([H])C(=C(C=2O1)[C@@]1([H])C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])[C@@]1([H])O[H])O[H])O[H])=O.Cl[H]
|
|
| InChi Key |
LGMSNQNWOCSPIK-LWHGMNCYSA-N
|
|
| InChi Code |
InChI=1S/C21H20ClNO5.ClH/c1-23-7-6-12(17(27)10-23)19-14(24)8-15(25)20-16(26)9-18(28-21(19)20)11-4-2-3-5-13(11)22;/h2-5,8-9,12,17,24-25,27H,6-7,10H2,1H3;1H/t12-,17+;/m0./s1
|
|
| 化学名 |
2-(2-chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methylpiperidin-4-yl]chromen-4-one;hydrochloride
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2815 mL | 11.4077 mL | 22.8154 mL | |
| 5 mM | 0.4563 mL | 2.2815 mL | 4.5631 mL | |
| 10 mM | 0.2282 mL | 1.1408 mL | 2.2815 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00098579 | Completed | Drug: doxorubicin hydrochloride Drug: alvocidib |
Gastrointestinal Stromal Tumor Stage IV Adult Soft Tissue Sarcoma |
National Cancer Institute (NCI) |
October 2004 | Phase 1 |
| NCT00047307 | Completed | Drug: gemcitabine hydrochloride Drug: alvocidib |
Stage IV Pancreatic Cancer Stage III Pancreatic Cancer |
National Cancer Institute (NCI) |
August 2002 | Phase 1 |
| NCT00407966 | Completed | Drug: alvocidib Drug: cytarabine |
Adult Acute Monocytic Leukemia (M5b) Adult Erythroleukemia (M6a) |
National Cancer Institute (NCI) |
October 2006 | Phase 2 |
| NCT01349972 | Completed | Drug: alvocidib Drug: cytarabine |
Adult Acute Monocytic Leukemia (M5b) Adult Erythroleukemia (M6a) |
National Cancer Institute (NCI) |
April 2011 | Phase 2 |
| NCT00470197 | Completed | Drug: alvocidib Drug: cytarabine |
Adult Erythroleukemia (M6a) Malignant Neoplasm |
National Cancer Institute (NCI) |
April 2007 | Phase 1 |
 HCl") Effect of FVP on the expression of PRGs and IRGs in BJ-TERT fibroblasts.Cell Div.2012 Mar 29;7:11. doi: 10.1186/1747-1028-7-11. |
|---|
 HCl") Biphasic effect of FVP in the expression of certain PRG/IRGs.Cell Div.2012 Mar 29;7:11. doi: 10.1186/1747-1028-7-11. |
 HCl") Certain PRG/IRGs are significantly expressed in the presence of FVP during a mitogenic response.Cell Div.2012 Mar 29;7:11. doi: 10.1186/1747-1028-7-11. |
 M1-20
M1-20
 CDK12-Cyclin K Ligand-Linker Conjugates 1
CDK12-Cyclin K Ligand-Linker Conjugates 1
 CGP-74514 dihydrochloride
CGP-74514 dihydrochloride
 (S)-CDK2 degrader 6
(S)-CDK2 degrader 6
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
