| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
Human PNP (IC50 = 1.19 nM); Mouse PNP (IC50 = 0.48 nM); Rat PNP (IC50 = 1.24 nM); Monkey PNP (IC50 = 0.66 nM); Dog PNP (IC50 = 1.57 nM)[2]
|
|---|---|
| 体外研究 (In Vitro) |
呋咯地辛 (10-30 μM) 处理 24 和 48 小时可部分减少 RPMI-8226、MOLT-4 和 5T33MM 细胞的增殖 [1]。 MM 细胞不受呋咯地辛影响(10-30 μM;24 和 48 小时;RPMI-8226、MOLT-4 和 5T33MM 细胞);然而,呋咯地辛会使 MOLT-4 细胞中活细胞的百分比降低 40%[1]。 Forodesine (BCX-1777) 可抑制各种药物、混合淋巴细胞反应 (MLR) 和白细胞介素 2 (IL-2) 刺激的人淋巴细胞增殖。植物血凝素 (PHA) 的 IC50 值小于 0.1-0.38 μM [2]。
多发性骨髓瘤(MM)是第二常见的血液系统恶性肿瘤,其特征是骨髓中恶性细胞的单克隆增殖。尽管最近在治疗策略方面取得了进展,但多发性骨髓瘤仍然无法治愈,需要新的治疗靶点。最近发现,嘌呤核苷磷酸化酶抑制剂呋咯地辛通过增加dGTP水平诱导慢性淋巴细胞白血病患者的白血病细胞凋亡。因此,我们测试了呋咯地辛是否能够通过类似的途径抑制小鼠和人类MM细胞的增殖和/或诱导凋亡。我们发现,在用呋咯地辛治疗48小时后,5T33MM和RPMI-8226 MM细胞中的dGTP略有增加,这与部分增殖抑制和有限的凋亡诱导有关。在研究导致细胞周期阻滞和凋亡的途径时,我们观察到p27、胱天蛋白酶3和BIM的上调。我们可以得出结论,呋咯地辛对MM细胞有一定的影响,但不如已知的对白血病细胞的影响那么显著。然而,呋咯地辛可能对MM中其他已建立的细胞毒性药物具有增强作用[2]。 PNP酶抑制[2] BCX-1777非常有效地抑制了人、小鼠、大鼠、猴子和狗的PNP。IC50值范围为0.48至1.57 nM(表1)。所有这些酶在3-10nM时的最大抑制效果约为90%至100%,表明BCX-1777是人类、小鼠、大鼠、猴子和狗PNP的强效抑制剂。 BCX-1777对IL-2、MLR-和PHA刺激的人T细胞淋巴增殖的抑制作用[2] 我们研究了BCX-1777抑制IL-2诱导的正常供体人PBL增殖的能力,结果如图1A所示。在dGuo(10μM)存在下,添加BCX-1777对IL-2的增殖反应产生了剂量依赖性抑制,IC50约为0.06μM。在没有dGuo的情况下,BCX-1777的IC50大于100μM。单独使用10μM的dGuo对活化淋巴细胞的增殖没有抑制作用。作为与同种异体排斥反应相关的体外实验,我们评估了BCX-1777在混合淋巴细胞反应中抑制四个供体对同种异体抗原刺激的人PBL增殖反应的能力(图1B)。如图1B所示,BCX-1777对这种反应产生了剂量依赖性的抑制作用,在0.05μM时抑制率为50%,在1μM时,抑制率为90-100%。在没有dGuo的情况下,BCX-1777在100μM以下没有明显的抑制作用。还检查了BCX-1777抑制PHA刺激的4名正常供体PBL增殖的能力(图1C)。在10μM dGuo存在下,BCX-1777对PHA刺激的细胞表现出剂量依赖性抑制作用,IC50为0.387μM。在没有dGuo的情况下,没有观察到明显的抑制作用。与未受刺激的淋巴细胞相比,用IL-2和PHA刺激的人淋巴细胞的胸苷掺入量增加了20倍以上,而用MLR刺激时,胸苷掺合量增加了10倍以上。 T细胞中的核苷酸库[2] 在BCX-1777和10μM dGuo存在的情况下,IL-2刺激的PBL显示出dGTP的积累(表2)。与对照样品相比,0.1μM BCX-1777的dGTP大约增加了4.5倍,1μM BCX-1777的dGTP增加了7.5倍。dGTP的增加与IL-2刺激的T细胞增殖的抑制平行。dGTP增加4.5倍产生52%的抑制作用,dGTP增加7.5倍产生76%的抑制作用。脱氧胞苷抑制激酶介导的dGuo转化为dGMP。为了进一步评估dGTP在T细胞抑制中的作用,在BCX-1777、dGuo和脱氧胞苷存在的情况下进行了增殖和核苷酸分析。脱氧胞苷(3μM)部分逆转了BCX-1777和dGuo对T细胞增殖的抑制作用。用BCX-1777、dGuo和dCyt孵育的T细胞的核苷酸库的测定显示,与用BCX-1777和dGuo孵育细胞相比,dGTP降低。不使用更高浓度(≥10μM)的脱氧胞苷,因为它会抑制T细胞的增殖。 IL-2刺激小鼠脾细胞增殖[2] 为了评估BCX-1777的特异性,我们评估了BCX-1777在存在和不存在dGuo(10μM)的情况下抑制IL-2诱导的小鼠脾细胞增殖的能力。在存在和不存在dGuo(10μM)的情况下,在高达100μM的BCX-1777浓度下,均未观察到对IL-2诱导的小鼠脾细胞增殖的显著抑制。与人类淋巴细胞不同,在BCX-1777(1μM)和dGuo(10μM)存在的情况下,小鼠脾细胞没有表现出dGTP的积累。 3.5.口服生物利用度和体内药理活性[2] 口服和静脉注射BCX-1777对血浆水平的影响结果如图3A所示。口服BCX-1777后迅速吸收,半小时内达到约3μM的Cmax。3小时时,血浆药物水平为1.1μM,6小时时检测不到药物。经计算,BCX-1777在小鼠体内的口服生物利用度为63%。 |
| 体内研究 (In Vivo) |
Forodesine (BCX-1777) 在小鼠体内具有出色的口服生物利用度 (63%) [2]。在小鼠中,单剂量 10 mg/kg 的 Forodesine 将 dGuo 升高至约 5 μM [2]。在人外周血淋巴细胞严重联合免疫缺陷(hu-PBL-SCID)小鼠模型中,呋咯地辛可有效延长寿命2倍以上[2]。
小鼠T细胞模型[2] 在DNFB诱导的接触性迟发型超敏反应小鼠耳水肿模型中评估了呋咯地辛/BCX-1777的体内疗效。迟发型超敏反应是由T细胞介导的。BCX-1777在30mg/kg剂量下对减轻小鼠耳水肿无效。环孢菌素(50mg/kg)用作阳性对照,耳肿胀减少58%。BCX-1777也在移植物抗宿主(GVH)诱导的小鼠脾肿大模型中进行了评估。通过注射对同种异体MHC抗原有反应并诱导全身GVHR症状的亲本T细胞,可以在成年F1小鼠中诱导实验性移植物抗宿主反应(GVHR)。GVHR类似于由T细胞对受体组织的细胞表面抗原反应介导的DTH反应。环孢菌素显著抑制了53%的GVH诱导的脾肿大,而BCX-1777在这种小鼠T细胞模型中无效。 Hu-PBL SCID小鼠模型[2] Mosier等人报道了将人PBL移植到SCID小鼠体内以构建hu-PBL SCID小鼠,并建议将其作为研究正常人类免疫功能的有用模型。最近,Sandhu等人[19]描述了一种将hu-PBLs有效植入SCID小鼠的新方案。移植到这些SCID小鼠体内的hu-PBL被证明可以通过诱导人类初级反应发挥作用。该方案的植入效率非常高,几乎100%的hu-PBL SCID小鼠在注射人PBL后不到4周内死于异种移植物抗宿主病(XGVHD)。在SCID小鼠中植入hu-PBL可诱导严重的XGVHD,并伴有体重减轻、腹泻、驼背和皮毛褶皱。这些小鼠最终因人类淋巴细胞浸润SCID小鼠组织而死亡。为了确定呋咯地辛/BCX-1777是否抑制小鼠体内人类T细胞的增殖并影响这些小鼠的寿命,从植入人类PBL前5天开始,以20mg/kg/天(b.i.d.)的剂量口服BCX-1777治疗SCID小鼠。对动物进行药物预处理以保持dGuo水平升高。按照相同的时间表继续给药,直到动物死亡。将来自三个供体的人淋巴细胞移植到三组SCID小鼠(n=10)中。实验动物(n=5)用药物治疗,对照动物(n=5)用载体(0.5%CMC)治疗。图5显示了这些实验中存活小鼠与存活天数的代表性曲线。在一项实验中,BCX-1777处理的小鼠平均寿命为30天,而对照组为15天。在另外两个实验中,BCX-1777治疗的小鼠分别存活了39天和43天,而未治疗组分别存活了17天和20天。因此,在每个实验中,BCX-1777将这些小鼠的寿命延长了2倍或更多(表3) |
| 酶活实验 |
嘌呤核苷磷酸化酶(PNP)缺乏症患者表现出选择性T细胞免疫缺陷。因此,PNP抑制剂作为潜在的T细胞选择性免疫抑制剂引起了人们的兴趣。BCX-1777是一种来自人类、小鼠、大鼠、猴子和狗等不同物种的PNP强效抑制剂,IC50值在0.48至1.57 nM之间。BCX-1777在2'-脱氧鸟苷(dGuo,3-10微M)存在下,抑制由白细胞介素-2(IL-2)、混合淋巴细胞反应(MLR)和植物血凝素(PHA)等各种试剂激活的人类淋巴细胞增殖(IC50值<0.1-0.38微M)。BCX-1777是一种比PD141955和BCX-34等其他已知PNP抑制剂强10-100倍的人类淋巴细胞增殖抑制剂。人淋巴细胞的核苷酸分析表明,BCX-1777对增殖的抑制与细胞中的dGTP水平相关[2]。
|
| 细胞实验 |
细胞增殖测定[1]
细胞类型:人 RPMI-8226、人 MOLT-4 (T-ALL) 细胞、5T33MM(多发性骨髓瘤,MM) 测试浓度: 10 μM、20 μM、30 μM 孵育时间: 24 和 48 小时 实验结果: 48 小时后(小时 )),MOLT-4细胞增殖被完全阻断,5T33MM细胞增殖减少15%。 细胞凋亡分析[1] 细胞类型:人 RPMI-8226、人 MOLT-4 (T-ALL) 细胞、5T33MM(多发性骨髓瘤,MM) 测试浓度:10 μM、20 μM、30 μM 孵育持续时间:24 小时和 48 小时(hrs) 实验结果: 细胞凋亡的诱导有限。 |
| 动物实验 |
BCX-1777 在小鼠体内具有极佳的口服生物利用度(63%)。小鼠单次服用 10 mg/kg 剂量的 BCX-1777 后,dGuo 水平可升高至约 5 μM。由于小鼠 T 细胞不像人 T 细胞那样会积累 dGTP,因此 BCX-1777 在小鼠 T 细胞模型(例如迟发型超敏反应 (DTH) 和脾肿大)中无效。然而,在人外周血淋巴细胞重症联合免疫缺陷 (hu-PBL-SCID) 小鼠模型中,BCX-1777 可有效延长小鼠寿命 2 倍或以上。这是首个已知的 PNP 抑制剂,其在小鼠体内可使 dGuo 水平升高至与 PNP 缺陷患者相似的水平。此外,BCX-1777 体外抑制 T 细胞活性也需要达到这样的 dGuo 水平。因此,BCX-1777 代表了一类新型选择性免疫抑制剂,可能对多种 T 细胞疾病具有治疗价值。[2]
口服生物利用度和体内药理活性[2] 将四只雌性小鼠(Balb/c)分组,分别单次口服或静脉注射溶于无菌生理盐水中的药物。在不同时间点,对小鼠进行麻醉(吸入麻醉),并通过眼眶后静脉丛采血。每个时间点均使用不同的小鼠组。血液经离心后收集血浆,并储存于 -20°C 直至分析。观察动物 48 小时,记录其死亡率和发病率。采用反相高效液相色谱法测定血浆药物浓度,定量限为 0.5 μM。采用反相高效液相色谱法测定脱氧鸟苷浓度,该分析的定量限为 0.75 μM。采用非房室模型和Winnonlin软件计算口服生物利用度。 小鼠T细胞模型[2] 小鼠迟发型超敏反应(DTH)的实验方法参照Braida和Knop以及Walsh等人的描述。简而言之,在第0天和第1天(给药后1小时),将25 μl 0.5% 2,4-二硝基氟苯(DNFB)的丙酮/橄榄油(4:1)溶液涂抹于小鼠剃毛后的腹部皮肤上进行致敏。小鼠每天(qd)分别给予赋形剂、福罗地辛/BCX-1777(30 mg/kg,口服)或环孢素A(50 mg/kg,腹腔注射),连续6天。在第5天,将20 μl 0.3% DNFB丙酮/橄榄油(4:1)溶液涂抹于小鼠右耳,24小时后,用游标卡尺测量耳廓厚度。 根据Roudebush和Bryant描述的方法,通过腹腔注射亲代品系(C57BL/6)的脾细胞,测量雄性杂交小鼠脾肿大的程度,来评估移植物抗宿主(GVH)反应。简而言之,从C57BL/6小鼠中无菌取出脾脏,并在聚丙烯网筛中进行分离。用氯化铵缓冲液裂解后,脾细胞用改良的HBSS洗涤两次,并在第0天以0.5 ml/只的体积腹腔注射(2×10⁸个细胞/ml)至杂交小鼠体内。杂交小鼠分别接受BCX-1777(30 mg/kg,口服)或环孢素A(50 mg/kg,腹腔注射)治疗10天;对照组小鼠在第0天接受HBSS或亲代脾脏注射,并分别腹腔注射或口服载体。取出脾脏并称重,结果以脾脏重量与体重的比值记录。 Hu-PBL SCID小鼠模型[2] 按照Sandhu等人描述的方法,将人外周血淋巴细胞(hu-PBL)移植到重症联合免疫缺陷(SCID)小鼠体内。在注射人外周血淋巴细胞(hu-PBL)前1天,SCID小鼠预先注射单剂量抗ASGM1抗体。抗ASGM1抗体为兔多克隆抗体,可识别小鼠NK细胞并抑制NK细胞活性。在hu-PBL移植前,SCID小鼠接受照射。照射剂量为3 Gy,使用137Cs源。用于移植到SCID小鼠体内的人类淋巴细胞分离自志愿者供体的白细胞层或全血。hu-PBL通过Ficoll-Hypaque密度梯度离心法分离,并在无菌条件下腹腔注射(每只小鼠注射3.0–5.0×10⁷个PBL)到SCID小鼠体内。实验动物预先接受福洛地辛/BCX-1777(20 mg/kg/天,每日两次)溶于0.5%羧甲基纤维素(CMC)的溶液处理5天,以提高dGuo水平。对照组动物仅接受0.5% CMC处理。比较对照组和实验组动物的寿命。 |
| 药代性质 (ADME/PK) |
口服生物利用度和体内药理活性[2]
图3A显示了BCX-1777口服和静脉给药后血浆浓度的变化。BCX-1777口服后吸收迅速,半小时内达到约3 μM的血药浓度峰值(Cmax)。3小时后,血浆药物浓度为1.1 μM,6小时后则检测不到。计算得出小鼠BCX-1777的口服生物利用度为63%。 在小鼠口服BCX-1777(10 mg/kg)后,监测了血浆dGuo水平的变化(图3B)。dGuo随时间增加,3小时时达到约5 μM的血药浓度峰值(Cmax)。6小时时,dGuo水平约为2.2 μM。24小时后,dGuo检测不到。血浆dGuo水平与药物浓度并非完全平行。 dGuo 达到血药浓度峰值 (Cmax) 的时间(3 小时)比药物达到血药浓度峰值 (Cmax) 的时间(约 30 分钟)存在延迟。在小鼠口服不同剂量 BCX-1777 3 小时后,监测血浆中 dGuo 和药物的浓度(图 4)。BCX-1777 可使 dGuo 水平呈剂量依赖性升高,直至 10 mg/kg 剂量。超过此剂量后,dGuo 水平不再升高。然而,血浆药物浓度随着 BCX-1777 剂量的增加而升高,直至 100 mg/kg。 |
| 参考文献 | |
| 其他信息 |
Immucillin H 是一种吡咯并嘧啶类和二羟基吡咯烷类化合物。
福洛地辛是一种高效、口服有效的、经合理设计的嘌呤核苷磷酸化酶 (PNP) 抑制剂,在临床前研究中已显示出对恶性细胞的活性,并在临床上对 T 细胞急性淋巴细胞白血病和皮肤 T 细胞淋巴瘤具有疗效。其他初步研究结果支持其用于治疗某些 B 细胞恶性肿瘤。 福洛地辛是一种嘌呤核苷磷酸化酶 (PNP) 的过渡态类似物抑制剂,具有潜在的抗肿瘤活性。给药后,福洛地辛优先结合并抑制 PNP,导致脱氧鸟苷三磷酸的积累,进而抑制核糖核苷二磷酸还原酶和 DNA 合成。该药物可选择性地诱导受刺激或恶性T淋巴细胞凋亡。 药物适应症 已研究用于治疗淋巴瘤(非霍奇金淋巴瘤)和白血病(淋巴样白血病)。 嘌呤核苷磷酸化酶 (PNP) 缺乏症患者表现出选择性T细胞免疫缺陷。因此,PNP抑制剂作为潜在的T细胞选择性免疫抑制剂备受关注。BCX-1777是一种强效的PNP抑制剂,对包括人、小鼠、大鼠、猴和犬在内的多种物种均有效,其IC50值范围为0.48至1.57 nM。在 2'-脱氧鸟苷 (dGuo,3-10 μM) 存在的情况下,BCX-1777 可抑制由多种因子激活的人淋巴细胞增殖,例如白细胞介素-2 (IL-2)、混合淋巴细胞反应 (MLR) 和植物血凝素 (PHA)(IC50 值 < 0.1-0.38 μM)。BCX-1777 对人淋巴细胞增殖的抑制作用比其他已知的 PNP 抑制剂(如 PD141955 和 BCX-34)强 10-100 倍。人淋巴细胞的核苷酸分析表明,BCX-1777 对细胞增殖的抑制作用与细胞内 dGTP 水平相关。BCX-1777 在小鼠体内具有良好的口服生物利用度 (63%)。小鼠单次服用 10 mg/kg 剂量的 BCX-1777 后,dGuo 可升高至约 5 μM。 BCX-1777 在小鼠 T 细胞模型(例如迟发型超敏反应 (DTH) 和脾肿大)中无效,因为小鼠 T 细胞不像人类 T 细胞那样会积累 dGTP。然而,在人外周血淋巴细胞重症联合免疫缺陷 (hu-PBL-SCID) 小鼠模型中,BCX-1777 可有效延长小鼠寿命 2 倍或更多。这是首个已知的 PNP 抑制剂,其可使小鼠体内 dGuo 水平升高至与 PNP 缺陷患者相似的水平。此外,这些 dGuo 水平也是 BCX-1777 体外抑制 T 细胞所必需的。因此,BCX-1777 代表了一类新型的选择性免疫抑制剂,可能对多种 T 细胞疾病具有治疗价值。[2] 总之,BCX-1777 是一种有效的 PNP 酶抑制剂,可抑制人类 T 细胞增殖。 BCX-1777 在小鼠体内具有口服生物利用度,能够最大程度地抑制 PNP,从而提高 dGuo 水平。这种 dGuo 水平的升高在 PNP 缺陷患者中发现,并已被证实是 T 细胞抑制所必需的。利用 hu-PBL SCID 小鼠模型,证实了 BCX-1777 的体内疗效。鉴于体外和体内数据,我们得出结论:BCX-1777 是一种新型的、口服有效的、T 细胞选择性免疫抑制剂,可用于治疗 T 细胞增殖性疾病。[2] |
| 分子式 |
C11H14N4O4
|
|---|---|
| 分子量 |
266.25326
|
| 精确质量 |
266.102
|
| 元素分析 |
C, 49.62; H, 5.30; N, 21.04; O, 24.04
|
| CAS号 |
209799-67-7
|
| 相关CAS号 |
Forodesine hydrochloride;284490-13-7
|
| PubChem CID |
135409409
|
| 外观&性状 |
Light brown to gray solid
|
| 密度 |
2.01
|
| 沸点 |
613.5ºC at 760mmHg
|
| 闪点 |
324.8ºC
|
| 蒸汽压 |
6.61E-16mmHg at 25°C
|
| 折射率 |
1.894
|
| LogP |
-2.3
|
| tPSA |
134.26
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
19
|
| 分子复杂度/Complexity |
404
|
| 定义原子立体中心数目 |
4
|
| SMILES |
O=C1C(NC=C2[C@@H]3N[C@H](CO)[C@@H](O)[C@H]3O)=C2NC=N1
|
| InChi Key |
IWKXDMQDITUYRK-KUBHLMPHSA-N
|
| InChi Code |
InChI=1S/C11H14N4O4/c16-2-5-9(17)10(18)7(15-5)4-1-12-8-6(4)13-3-14-11(8)19/h1,3,5,7,9-10,12,15-18H,2H2,(H,13,14,19)/t5-,7+,9-,10+/m1/s1
|
| 化学名 |
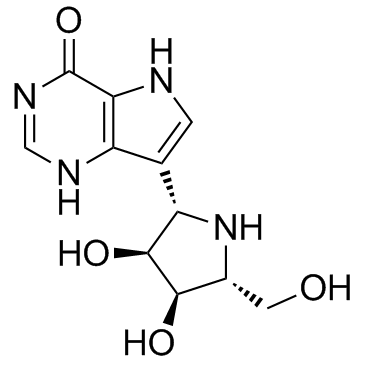
7-[(2S,3S,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)pyrrolidin-2-yl]-3,5-dihydropyrrolo[3,2-d]pyrimidin-4-one
|
| 别名 |
Forodesine; Immucillin H; 209799-67-7; Fodosine; Immucillin-H; mundesine; BCX-1777; 1,4-dideoxy-4-aza-1-(s)-(9-deazahypoxanthin-9-yl)-d-ribitol; Immucillin H; NTR 001; NTR-001; NTR001
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~375.59 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (9.39 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (9.39 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (9.39 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.7559 mL | 18.7793 mL | 37.5587 mL | |
| 5 mM | 0.7512 mL | 3.7559 mL | 7.5117 mL | |
| 10 mM | 0.3756 mL | 1.8779 mL | 3.7559 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00646165 | Terminated | Drug: Forodesine | B-cell Chronic Lymphocytic Leukemia | Mundipharma Research Limited | July 2008 | Phase 1 |
| NCT00742495 | Terminated | Drug: Forodesine | Relapsed or Refractory T-cell Acute Lymphoblastic Leukaemia |
Mundipharma Research Limited | March 2009 | Phase 1 Phase 2 |
| NCT00501735 | Completed | Drug: Forodesine 200 mg | Cutaneous T-cell Lymphoma (CTCL), | BioCryst Pharmaceuticals | July 2007 | Phase 2 |
| NCT00289549 | Completed | Drug: forodesine hydrochloride (BCX-1777) |
Leukemia, Lymphocytic, Chronic | BioCryst Pharmaceuticals | June 2005 | Phase 2 |
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1



































 463611831
463611831
