| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
RIP1 kinase (IC50 = 29 nM)
GSK-963 targets receptor-interacting protein 1 (RIP1) kinase (IC₅₀ = 1–4 nM for inhibiting RIP1-dependent cell death in human and murine cells; >10,000-fold selective for RIP1 over 339 other human kinases at 10 μM; no measurable activity against indoleamine-2,3-dioxygenase (IDO)) [1] |
|---|---|
| 体外研究 (In Vitro) |
GSK-963 抑制人和小鼠细胞中 RIP1 依赖性细胞死亡,IC50 范围为 1 至 4 nM[1]。
GSK′963在生化检测中是RIP1激酶的强效选择性抑制剂。 GSK′963是一种选择性强的小鼠和人类细胞体外坏死性下垂抑制剂。 1. GSK-963在荧光偏振(FP)结合实验中对RIP1的ATP结合口袋表现出极强的结合亲和力,远高于坏死抑制剂1(Nec-1);在ADP-Glo激酶实验中,其可在体外强效抑制RIP1激酶域的自磷酸化,效能显著优于Nec-1 [1] 2. 在基于细胞的坏死性凋亡实验中,GSK-963以1–4 nM的IC₅₀抑制TNF + zVAD诱导的小鼠纤维肉瘤L929细胞、人单核细胞U937、原代小鼠骨髓来源巨噬细胞(BMDM)的坏死性凋亡;在原代人中性粒细胞中(用TNF + zVAD + SMAC模拟物诱导坏死性凋亡),其抑制活性的IC₅₀同样为1–4 nM;而其非活性对映体GSK-962在相同浓度下无抑制作用 [1] 3. GSK-963(100 nM)对TNF + 环己酰亚胺处理的BMDM中caspase 3/7的活性无影响(该模型用于诱导细胞凋亡),也不抑制TNF诱导的BMDM中IκB的磷酸化(5分钟时)和降解(15分钟时),表明其不干扰凋亡信号通路或NF-κB的激活 [1] 4. GSK-963在体外酶活实验中对IDO无抑制活性(以甲萘醌为IDO抑制的阳性对照);在10 μM浓度下,对339种测试的人类激酶均无抑制作用 [1] |
| 体内研究 (In Vivo) |
GSK-963 是 TNF+zVAD 引起的致命性休克的强效抑制剂。与 Nec-1 相比,GSK-963 在 2 mg/kg 剂量下可在较长时间内将血液浓度维持在抑制 RIP1 活性 90% 所需的水平之上。
GSK′963是TNF+zVAD介导的致死性休克的强效抑制剂:研究人员评估了GSK′965和Nec-1在体内腹腔注射后的药代动力学特征。尽管GSK′963的表观半衰期大于Nec-1(图3a;补充图1a),但Nec-1在10mg/kg时的暴露量比葛兰素史克′963高出约10倍。然而,基于小鼠药代动力学特征(图3a;补充图1a)和TNF+zVAD处理的L929细胞中的化合物效力(图2a)对这两种化合物的药效学建模表明,与Nec-1相比,在2 mg/kg的剂量下,GSK′963将在较长一段时间内将血液浓度维持在90%抑制RIP1活性所需的浓度以上(图3b;补充图1b)。为了直接在体内测试GSK′963的疗效,我们使用了无菌休克的急性模型。给予TNF+zVAD会导致致命的低体温,此前已被证明这取决于RIP1激酶活性。5,18用2 mg/kg GSK′963治疗动物可完全保护动物免受TNF+zVASD诱导的体温下降,0.2 mg/kg剂量也显示出显著的反应(图3c)。正如预期的那样,GSK′962对TNF+zVAD诱导的休克没有影响,证实GSK′963通过抑制RIP1激酶选择性地起作用(图3d)。有趣的是,Nec-1在0.2 mg/kg的剂量下对模型没有影响,该剂量在文献中通常用于在体内抑制RIP1,并且在高10倍的剂量下显示出最低水平的保护作用(图3e)。这些结果共同表明,与Nec-1相比,GSK′963是探索体内急性RIP1激酶生物学的更好工具分子[1]。 1. 在TNF + zVAD诱导的小鼠无菌休克模型中,C57BL/6小鼠腹腔预处理GSK-963(0.2 mg/kg或2 mg/kg)15分钟后,再静脉注射TNF + zVAD,可在3小时的监测期内显著保护小鼠免于体温降低;该保护作用呈剂量依赖性,且效果远优于同剂量(0.2 mg/kg和2 mg/kg)的Nec-1 [1] 2. GSK-963的非活性对映体GSK-962即使在20 mg/kg的高剂量下(腹腔注射),也无法对TNF + zVAD诱导的体温降低产生保护作用,证实了GSK-963对RIP1的靶点特异性作用 [1] 3. 药代动力学建模显示,C57BL/6小鼠腹腔注射10 mg/kg GSK-963后产生的血浆药物暴露量,足以在0.2 mg/kg和2 mg/kg剂量下实现对RIP1的有效抑制(基于其在L929细胞中的效能数据)[1] |
| 酶活实验 |
使用 ADP-Glo 发光测定法测定化合物对抗 RIP1 激酶活性的效力,该测定法测量 ATP 到 ADP 的转化,如前所述。简而言之,主要反应由 50 mM HEPES pH 7.5、50 mM NaCl、30 mM MgCl2、1 mM DTT、0.5 mg/ml BSA 和 0.02% 中的 10 nM GST-RIPK1 (1–375) 和 50 μM ATP 组成章节。将 5 微升酶和 5 μl ATP 以最终测定浓度的两倍添加到板中,并在室温下孵育 4 小时。在读板器上测量发光。测试化合物抑制表示为内部测定对照的抑制百分比。
荧光偏振(FP)结合分析[1] 如前所述,开发了一种基于FP的结合测定法,通过与荧光标记的ATP竞争配体竞争,定量RIP1 ATP结合袋处新型测试化合物的相互作用。18简而言之,GST-RipK1(1-375)被纯化,并以10nM的最终测定浓度使用。使用荧光标记配体(14-(2-{[3-({2-{[4-(氰甲基)苯基]氨基}-6-[(5-环丙基-1H-吡唑-3-基)氨基]-4-嘧啶基}氨基)丙基]氨基}-2-氧乙基)-16,16,18,18-四甲基-6,7,7a,8a,9,10,16,18-八氢苯并[2“,3”]中氮茚并[8“,7”:5ʹ,6ʹ;]吡喃并[3,2ʹ:3,4]吡啶并[1,2-A]吲哚-5-鎓-2-磺酸盐,最终测定浓度为5nM。样本在Analyst多模式阅读器上读取。试验化合物抑制率表示为内部试验对照的抑制百分比(%)。 ADP-Glo激酶测定[1] 使用ADP-Glo发光测定法测定化合物对RIP1激酶活性的效力,该测定法测量ATP向ADP的转化,如前所述。18简而言之,主要反应由10 nM GST-RIPK1(1-375)和50μM ATP在50 mM HEPES pH 7.5、50 mM NaCl、30 mM MgCl2、1 mM DTT、0.5 mg/ml BSA和0.02%CHAPS中组成。将5微升酶和5μl ATP以最终测定浓度的两倍加入平板中,在室温下孵育4小时。在平板读数器上测量发光。试验化合物抑制率表示为内部测定对照的抑制百分比。 激酶选择性[1] 在Reaction Corp Biology使用P33放射性标记测定法对GSK′963进行了抗339种激酶的测试(http://www.reactionbiology.com). 该化合物在10μM下以单次剂量进行了两次测试。反应在10μM ATP下进行。数据以酶活性百分比报告(相对于DMSO对照)。完整数据集见补充表一。 IDO酶法测定[1] 如Takahashi等人25所述,使用购自R&D Systems的重组人IDO测定IDO活性。使用Spectramax酶标仪在490 nm下测量犬尿氨酸衍生的黄色色素。 半胱天冬酶3/7测定[1] 为了诱导细胞凋亡,用GSK′963(100 nM)、GSK′962(100 nM)或Nec-1(10μM)预处理30分钟的BMDM用TNF(50 ng/ml)和CHX(12μg/ml)刺激。使用Caspase-Glo 3/7测定法在6小时时测量Caspase 3/7活性。 1. 荧光偏振(FP)结合实验:将重组RIP1蛋白与系列浓度的GSK-963、GSK-962及Nec-1共同孵育,评估化合物与RIP1的ATP结合口袋的结合亲和力。检测荧光偏振信号并绘制剂量-响应曲线(GSK-963 n=7、GSK-962 n=4、Nec-1 n=52),比较各化合物的结合效能 [1] 2. ADP-Glo激酶实验:将重组RIP1激酶域与不同浓度的GSK-963、GSK-962或Nec-1在ATP存在下孵育,评估对RIP1自磷酸化的抑制作用。孵育后加入ADP-Glo试剂检测生成的ADP量(激酶活性的标志物),绘制剂量-响应曲线(GSK-963 n=2、GSK-962 n=2、Nec-1 n=20),并计算对RIP1激酶抑制的IC₅₀值 [1] 3. 激酶选择性实验:在10 μM浓度下,用GSK-963测试339种人类激酶的活性,检测各激酶的活性并计算抑制率,结果显示GSK-963对这些激酶均无抑制作用 [1] 4. IDO酶活实验:将GSK-963、GSK-962、Nec-1及阳性对照甲萘醌与IDO酶和其底物在体外体系中孵育,检测IDO的酶活性,实验重复三次,结果证实GSK-963无可检测的IDO抑制活性 [1] |
| 细胞实验 |
对于免疫印迹分析,用 GSK'963 (100 nM)、GSK'962 (100 nM) 或 Nec-1 (10 μM) 预处理 30 分钟后,用 50 ng/ml TNF 刺激 BMDM 5 和 15 分钟。在 1× 细胞裂解缓冲液中用蛋白酶和磷酸酶抑制剂制成的裂解物在 4-12% SDS-PAGE 上分离,并印迹到硝酸纤维素膜上。 IκB、磷酸-IκB 和微管蛋白作为印迹上的上样对照进行探测。
细胞培养[1] 将小鼠纤维肉瘤L929细胞(ATCC#CCL-1)和人单核细胞U937细胞在添加了10%热灭活FBS、100 U/ml青霉素和100 U/ml链霉素的RPMI培养基中培养。通过用10ng/ml M-CSF分化7天,从C57BL/6小鼠制备BMDM,并在添加了10%热灭活FBS、100U/ml青霉素、100μg/ml链霉素和0.25μg/ml两性霉素的DMEM培养基中培养。按照标准方法从人血液中分离出原代人中性粒细胞,该方法包括在右旋糖酐中顺序沉淀、在Ficoll-Hypaque中密度离心和溶解污染的红细胞。 基于细胞的检测[1] 在半胱氨酸天冬氨酸蛋白酶抑制剂zVAD FMK或QVD Oph的存在下,TNF诱导BMDM、L929和U937细胞发生坏死性细胞死亡(BMDM:50 ng/ml TNF+50μM zVAD;L929:100ng/ml肿瘤坏死因子+50μM QVD;U937:100 ng/ml TNF+25μM QVD)。为了评估RIP1抑制剂的效果,用化合物(剂量反应)预处理细胞30分钟。19-21小时后,通过使用CellTiter-Glo发光细胞活力测定法测量细胞ATP水平来评估诱导的细胞死亡。为了诱导中性粒细胞坏死性下垂,用TNF(10ng/ml)、QVD-Oph(50μM)和SMAC模拟物(100nM)刺激新鲜分离的人中性粒细胞。如上所述评估诱导的细胞死亡。 1. 坏死性凋亡细胞活力实验(CellTiter-Glo法):将小鼠纤维肉瘤L929细胞、人单核细胞U937、原代小鼠BMDM及原代人中性粒细胞接种于培养板,加入系列浓度的GSK-963、GSK-962或Nec-1;用TNF + zVAD诱导坏死性凋亡(人中性粒细胞需联合SMAC模拟物),孵育后加入CellTiter-Glo试剂检测细胞活力,整合至少三次独立实验的数据绘制剂量-响应曲线,并计算坏死性凋亡抑制的IC₅₀值 [1] 2. Caspase 3/7活性实验:原代小鼠BMDM用TNF + 环己酰亚胺处理以诱导凋亡,同时加入GSK-963(100 nM)、GSK-962(100 nM)或Nec-1(10 μM)。处理3小时后用Caspase-Glo 3/7试剂检测caspase 3/7活性,20小时后用CellTiter-Glo法检测细胞活力;实验独立重复四次,结果显示GSK-963对凋亡信号无影响 [1] 3. IκB磷酸化与降解的免疫印迹实验:原代小鼠BMDM预先用GSK-963(100 nM)、GSK-962(100 nM)或Nec-1(10 μM)处理,再用TNF刺激,分别在5分钟(检测IκB磷酸化)和15分钟(检测IκB降解)收集细胞裂解液;通过免疫印迹实验,用磷酸化IκB、总IκB和微管蛋白(内参)的抗体进行检测;实验用四只不同小鼠的细胞重复,结果证实GSK-963不影响TNF诱导的NF-κB信号通路 [1] |
| 动物实验 |
C57BL/6 小鼠在静脉注射 TNF (1.25 mg/kg) 和 zVAD-FMK (16.7 mg/kg) 前 15 分钟,腹腔注射 Nec-1 (0.2 和 2 mg/kg)、GSK′963 (0.2 和 2 mg/kg) 或 GSK′962 (20 mg/kg)。
C57BL/6 小鼠药代动力学实验[1] 简而言之,每种化合物每组使用 n=3 只动物进行药代动力学研究。动物腹腔注射 GSK′963 或 7-Cl-O-Nec-1,药物配制成含 6% 11-β-羟丙基环糊精和 5% DMSO 的水溶液。在腹腔注射后不同时间点采集血样,并用水 1:1 稀释后储存于 -80 °C。 在生物分析之前,将样品解冻,并使用基于蛋白质沉淀的方法分离每种分析物。将所得上清液注入针对目标化合物检测优化的LC/MS/MS系统。数据以标准曲线分析确定的定量药物浓度表示。在这些优化条件下,两种化合物的典型定量下限均为1.00 ng/ml。 TNF+zVAD诱导的休克模型[1] 在静脉注射TNF(1.25 mg/kg)和zVAD-FMK(16.7 mg/kg)前15分钟,腹腔注射Nec-1(0.2和2 mg/kg)、GSK′963(0.2和2 mg/kg)或GSK′962(20 mg/kg)。通过直肠探针监测体温,持续3小时。 1. 药代动力学 (PK) 研究:C57BL/6 小鼠腹腔注射 GSK-963,剂量为 10 mg/kg。在预定时间点采集血样,分离血浆,并测定血浆中 GSK-963 的浓度。PK 曲线由三只独立动物的合并结果生成,并基于其在 L929 细胞试验中的效力对 RIP1 的预测抑制率进行建模 [1]。 2. TNF + zVAD 诱导的无菌性休克模型:将 C57BL/6 小鼠随机分为若干组(每组七只)。小鼠在静脉注射TNF + zVAD前15分钟,分别接受GSK-963(0.2 mg/kg或2 mg/kg,腹腔注射)、GSK-962(20 mg/kg,腹腔注射)、Nec-1(0.2 mg/kg或2 mg/kg,腹腔注射)或载体(对照组)预处理。使用直肠探针每30分钟监测一次直肠温度,持续3小时。该实验独立重复三次,并绘制各组的温度变化曲线[1]。 |
| 药代性质 (ADME/PK) |
接下来,研究人员评估了腹腔注射(ip)GSK′963和Nec-1在体内的药代动力学特征。在10 mg/kg剂量下,Nec-1的暴露量比GSK′963高约10倍,尽管GSK′963的表观半衰期长于Nec-1(图3a;补充图1a)。然而,基于小鼠药代动力学特征(图3a;补充图1a)和化合物在TNF+zVAD处理的L929细胞中的效力(图2a)的药效学模型表明,在2 mg/kg剂量下,与Nec-1相比,GSK′963能够更长时间地维持高于抑制RIP1活性90%所需浓度的血药浓度(图3b;补充图1b)。 [1]
1. 将GSK-963以10 mg/kg的剂量腹腔注射给C57BL/6小鼠,并在不同时间点测量该化合物的血浆浓度,以生成药代动力学曲线。[1] 2. 基于观察到的GSK-963药代动力学曲线及其抑制TNF + zVAD诱导的L929细胞坏死性凋亡的效力,计算了0.2 mg/kg和2 mg/kg剂量下RIP1的预测抑制率,证实这些剂量在体内可有效抑制RIP1。[1] |
| 参考文献 | |
| 其他信息 |
坏死性凋亡及其由RIP1激酶活性调控的信号通路正逐渐成为多种疾病炎症的关键驱动因素。通过使用RIP1激酶抑制剂坏死抑制剂Necrostatin-1 (Nec-1),人们对RIP1如何调控坏死性细胞死亡有了更深入的了解。Nec-1一直是探索RIP1激酶活性功能的变革性工具;然而,其应用受到一定限制,例如效力中等、对吲哚胺-2,3-双加氧酶(IDO)存在脱靶活性以及药代动力学性质较差。Nec-1的这些局限性促使人们致力于寻找研究RIP1功能的下一代工具,并最终发现了7-Cl-O-Nec-1 (Nec-1s)。Nec-1s具有更优的药代动力学性质,且不抑制IDO。本文描述了GSK′963的特性,GSK′963是一种手性小分子RIP1激酶抑制剂,其化学结构与Nec-1和Nec-1s均不同。在生化和细胞实验中,GSK′963的效力均显著高于Nec-1,对人源和鼠源细胞中RIP1依赖性细胞死亡的抑制IC50值介于1至4 nM之间。GSK′963对RIP1的选择性比对其他339种激酶高10000倍以上,对IDO无明显活性,且存在一种无活性的对映体GSK′962,可用于验证其靶向作用。GSK′963体外活性的增强也体现在体内,在TNF诱导的无菌性休克模型中,相同剂量的GSK′963对低温的保护作用远优于Nec-1。我们相信,GSK′963 代表了用于体外和体内研究 RIP1 功能的下一代工具,并有助于阐明我们目前对 RIP1 在疾病发病机制中作用的理解。[1]
在本研究中,我们对 GSK′963 进行了表征,GSK′963 是一种新型 RIP1 激酶小分子抑制剂,其结构与 Nec-1 和 Nec-1s 截然不同。GSK′963 的效力比 Nec-1 高 200 倍以上,对 RIP1 激酶活性具有极高的选择性,并且对 IDO 活性、TNF 介导的 NFκB 激活或细胞凋亡均无影响。GSK′963 是一种手性分子,因此可以使用其化学结构相同的非活性对映体作为阴性对照,以验证该抑制剂的靶向活性。此外,在TNF诱导的无菌性休克体内模型中,GSK′963在Nec-1在该模型中未显示出显著保护作用的剂量下,提供了完全的保护作用。综上所述,我们认为GSK′963代表了探索RIP1激酶介导的生物学作用的又一新一代工具。[1] 综上所述,我们发现了一种新型的、高效且选择性的RIP1激酶活性抑制剂。我们认为GSK′963代表了用于体外研究RIP1生物学的新一代工具抑制剂,与市售的坏死抑制剂相比具有显著优势。 1.GSK-963是一种手性小分子RIP1激酶抑制剂,其化学结构与Nec-1和Nec-1s(7-Cl-O-Nec-1)不同;它解决了 Nec-1 的局限性(效力中等、脱靶 IDO 抑制、药代动力学性质差)[1] 2. GSK-963 有一个无活性的对映体 (GSK-962),可用作阴性对照,以验证 RIP1 抑制在体外和体内实验中的靶向作用[1] 3. 坏死性凋亡和 RIP1 激酶介导的信号传导是各种疾病情况下炎症的关键驱动因素; GSK-963是一种新一代工具化合物,因其高效、选择性强且无脱靶活性,可用于研究RIP1在疾病发病机制中的作用[1] 4. GSK-963特异性抑制RIP1依赖性坏死性凋亡,而不影响凋亡信号通路(caspase 3/7活性)或NF-κB激活(IκB磷酸化/降解),表明其对RIP1激酶具有靶向特异性[1] |
| 分子式 |
C14H18N2O
|
|
|---|---|---|
| 分子量 |
230.31
|
|
| 精确质量 |
230.141
|
|
| 元素分析 |
C, 73.01; H, 7.88; N, 12.16; O, 6.95
|
|
| CAS号 |
2049868-46-2
|
|
| 相关CAS号 |
GSK962;2049872-86-6
|
|
| PubChem CID |
122703613
|
|
| 外观&性状 |
Light yellow to yellow solid powder
|
|
| 密度 |
1.06±0.1 g/cm3
|
|
| 沸点 |
342.8±45.0 °C
|
|
| LogP |
2.1
|
|
| tPSA |
32.7
|
|
| 氢键供体(HBD)数目 |
0
|
|
| 氢键受体(HBA)数目 |
2
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
17
|
|
| 分子复杂度/Complexity |
311
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
O=C(C(C)(C)C)N1C(C2C=CC=CC=2)CC=N1
|
|
| InChi Key |
NJQVSLWJBLPTMD-LBPRGKRZSA-N
|
|
| InChi Code |
InChI=1S/C14H18N2O/c1-14(2,3)13(17)16-12(9-10-15-16)11-7-5-4-6-8-11/h4-8,10,12H,9H2,1-3H3/t12-/m0/s1
|
|
| 化学名 |
2,2-dimethyl-1-[(3S)-3-phenyl-3,4-dihydropyrazol-2-yl]propan-1-one
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.5 mg/mL (10.85 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (10.85 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液添加到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.3420 mL | 21.7099 mL | 43.4197 mL | |
| 5 mM | 0.8684 mL | 4.3420 mL | 8.6839 mL | |
| 10 mM | 0.4342 mL | 2.1710 mL | 4.3420 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
GSK′963A is a potent and selective inhibitor of RIP1 kinase. (a) Chemical structures of GSK′963A (active analog), GSK′962A (inactive analog) and Necrostatin-1. (b) Dose–response curves for GSK′963, GSK′962 and Nec-1 in the FP binding assay evaluating the affinity of compounds for RIP1 (ATP-binding pocket).Cell Death Discov.2015 Jul 27;1:15009. |
|---|
 GSK′963A is highly potent in human and mouse cell-based assays and selective for inhibition of necroptosis. (a–d) Dose–response curves for GSK′963, GSK′962 and Nec-1 in cell-based assays.Cell Death Discov.2015 Jul 27;1:15009. |
 GSK′963A protects mice from TNF+zVAD-induced hypothermia. (a) Pharmacokinetic profile of GSK′963A dosed i.p. at 10 mg/kg in C57BL/6 mice. The data represent the combined results of the three independent animals. (b) Modeling of predicted % inhibition against RIP1 using the observed pharmacokinetic profile of GSK′963 in conjunction with the potency in inhibiting TNF+zVAD necroptosis in mouse L929 cells.Cell Death Discov.2015 Jul 27;1:15009. |
 RIPK1-IN-36
RIPK1-IN-36
 RIPK1-IN-30
RIPK1-IN-30
 RIPK3-IN-6
RIPK3-IN-6
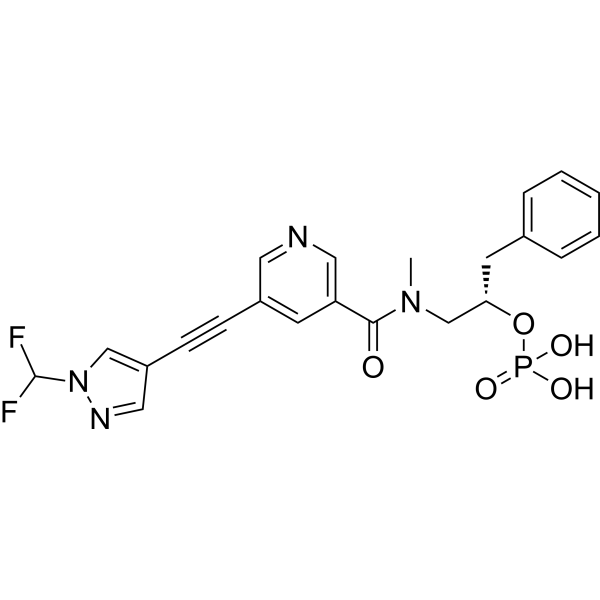 Fosizensertib
Fosizensertib
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
