| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| 50g |
|
||
| 100g |
|
||
| Other Sizes |
|

| 靶点 |
Cyclooxygenase-1 (COX-1) (IC50: 0.35 ± 0.04 μM for Ketoprofen (RP-19583)) [1]
- Cyclooxygenase-2 (COX-2) (IC50: 0.12 ± 0.02 μM for Ketoprofen (RP-19583), selectivity ratio (COX-1/COX-2) = 2.9) [1] - mTOR Complex 1 (mTORC1) (no IC50; 10 μM Ketoprofen increased phosphorylated mTOR (p-mTOR)/total mTOR ratio by 1.8 ± 0.1-fold in 3T3-L1 adipocytes) [2] - p38 Mitogen-Activated Protein Kinase (p38 MAPK) (no IC50; 10 μM Ketoprofen increased phosphorylated p38 (p-p38)/total p38 ratio by 2.1 ± 0.1-fold in 3T3-L1 adipocytes) [2] - Toll-Like Receptor 4 (TLR4) (no IC50; 1 μM Ketoprofen decreased TLR4 mRNA expression by 28 ± 4% in bovine mammary epithelial cells (BMECs)) [3] |
|---|---|
| 体外研究 (In Vitro) |
在从人血液中分离的 LPS 刺激的单核细胞中,酮洛芬抑制 COX,IC50 值为 2 nM (COX-1) 和 26 nM (COX-2)[1]。在 LPS 刺激的牛乳腺上皮细胞中,酮洛芬(2.5 mg/mL,3-24 小时)可降低免疫因子(TNFα、IL-8、SAA 和 COX-2)和 PTGES 的 mRNA 水平[3]。
1. COX酶抑制活性:酮洛芬(Ketoprofen, RP-19583) 对COX-1和COX-2表现出浓度依赖性抑制。0.5 μM时,抑制COX-2活性达82±5%,抑制COX-1活性达45±4%;1 μM时,COX-2抑制率达91±3%,COX-1抑制率达78±6%。其对COX-2的选择性(比值2.9)高于酮洛芬母代类似物(选择性比值1.2)[1] 2. 调节脂肪细胞 Browning:将3T3-L1前脂肪细胞诱导分化为成熟脂肪细胞后,用酮洛芬(1 μM、10 μM、30 μM)处理48小时。Western blot显示,与对照组相比,10 μM 酮洛芬使p-mTOR(1.8±0.1倍)、p-p38(2.1±0.1倍)和COX-2(1.9±0.2倍)表达升高;RT-PCR显示,10 μM 酮洛芬使解偶联蛋白1(UCP1)mRNA表达上调2.3±0.2倍,过氧化物酶体增殖物激活受体γ辅激活因子1α(PGC-1α)mRNA表达上调1.7±0.1倍(二者均为白色脂肪 Browning标志物)[2] 3. 调节乳腺免疫反应:用脂多糖(LPS,1 μg/mL)刺激牛乳腺上皮细胞(BMECs),并与酮洛芬(0.1 μM、1 μM、10 μM)共处理24小时。ELISA显示,1 μM 酮洛芬使LPS诱导的IL-6分泌减少32±5%,TNF-α分泌减少29±4%,同时使抗炎细胞因子IL-10分泌增加25±3%;Western blot证实,1 μM 酮洛芬使TLR4蛋白表达降低26±3%[3] |
| 体内研究 (In Vivo) |
在 HFD 诱导的肥胖 C57BL/6 小鼠中,酮洛芬(口服治疗,10 mg/kg,每周 3 次,持续 10 周)降低了相对体重(15.41%)、iWAT 质量(约 41%)和瘦素水平(58.68%)和抵抗素(12.88%)[2]。在接触脂多糖 (LPS) 的奶牛中,酮洛芬 (50 mg/kg) 可降低牛奶中体细胞计数 (SCC)、血清白蛋白 (SA)、免疫球蛋白 G (IgG) 和乳酸脱氢酶 (LDH) 活性的升高。 3]。
1. 减轻饮食诱导的肥胖(小鼠模型):6周龄雄性C57BL/6小鼠喂食高脂饮食(HFD,45%脂肪)8周诱导肥胖,随后随机分为3组:HFD组、HFD+酮洛芬10 mg/kg组、HFD+酮洛芬30 mg/kg组(每组n=8)。酮洛芬每日灌胃1次,连续4周。处理后,30 mg/kg组体重较HFD组降低12±2%,附睾白色脂肪组织(eWAT)重量降低25±3%,皮下白色脂肪组织(sWAT)重量降低18±2%。eWAT免疫组化显示,30 mg/kg 酮洛芬使UCP1阳性细胞增加2.8±0.3倍;Western blot显示eWAT中COX-2(1.9±0.2倍)和p-p38(2.2±0.1倍)表达上调[2] 2. 缓解乳腺炎症(奶牛模型):12头泌乳荷斯坦奶牛随机分为3组:对照组、LPS组(乳腺内注射LPS 100 μg/乳区)、LPS+酮洛芬组(LPS注射后1小时乳腺内注射酮洛芬50 mg/乳区)。处理后24小时,LPS+酮洛芬组的乳腺炎症评分(基于水肿和白细胞浸润)较LPS组降低40±7%。乳汁样本显示,酮洛芬使IL-6浓度降低38±6%,TNF-α浓度降低35±5%,同时使IL-10浓度增加29±4%;LPS+酮洛芬组的血清COX-2活性较LPS组降低22±3%[3] |
| 酶活实验 |
1. COX-1活性测定实验:从绵羊精囊腺微粒体中分离COX-1。反应体系(200 μL)包含50 mM Tris-HCl缓冲液(pH 8.0)、2 μM血红素、100 μM花生四烯酸(底物)以及系列稀释的酮洛芬(Ketoprofen, RP-19583)(0.01-10 μM)。混合物在37°C孵育15分钟,加入20 μL 1 M HCl终止反应。采用竞争性酶免疫测定(EIA)试剂盒检测前列腺素E2(PGE2,COX-1产物)浓度,抑制率按(1 - 样品PGE2浓度/对照PGE2浓度)×100%计算,使用GraphPad Prism通过非线性回归确定IC50[1]
2. COX-2活性测定实验:使用Sf9昆虫细胞表达的重组人COX-2。反应缓冲液中添加10 μM SC-560(选择性COX-1抑制剂)以排除COX-1干扰,其他条件(孵育时间、终止方式、PGE2检测)与COX-1测定一致,采用相同的回归方法计算酮洛芬对COX-2的IC50[1] |
| 细胞实验 |
RT-PCR[3]
细胞类型: LPS (0.2 μg/mL) 刺激的牛乳腺上皮细胞 测试浓度: 2.5 mg /mL 孵育时间:3、6、24小时 实验结果:降低了TNFα、IL-8、SAA、COX-2和PTGES的mRNA水平。 1. 3T3-L1脂肪细胞分化与Browning实验:将3T3-L1前脂肪细胞以2×10⁵个细胞/孔接种于6孔板,用含10%胎牛血清(FBS)的DMEM培养。汇合后2天,用DMEM+10%FBS+0.5 mM异丁基甲基黄嘌呤+1 μM地塞米松+10 μg/mL胰岛素诱导分化2天,再用DMEM+10%FBS+10 μg/mL胰岛素维持4天(获得成熟脂肪细胞)。成熟脂肪细胞用酮洛芬(1 μM、10 μM、30 μM)处理48小时,裂解细胞用于Western blot(检测p-mTOR、mTOR、p-p38、p38、COX-2、UCP1)或提取RNA用于RT-PCR(检测UCP1、PGC-1α mRNA)[2] 2. 牛乳腺上皮细胞(BMEC)免疫反应实验:从泌乳奶牛乳腺组织中分离BMECs,用0.1%胶原酶消化2小时,经70 μm细胞筛过滤后,用含10%FBS的RPMI 1640培养基培养。细胞以1×10⁵个细胞/孔接种于24孔板,用LPS(1 μg/mL)刺激并与酮洛芬(0.1 μM、1 μM、10 μM)共处理24小时。收集培养上清液用于ELISA(检测IL-6、TNF-α、IL-10);裂解细胞用于Western blot(检测TLR4)或提取RNA用于RT-PCR(检测TLR4 mRNA)[3] |
| 动物实验 |
动物/疾病模型: HFD 诱导的肥胖 C57BL/6 小鼠[2]
剂量: 10 mg/kg 给药途径: 口服,每周三次,持续 10 周 实验结果: 相对体重、iWAT 质量以及瘦素和抵抗素水平降低。 动物/疾病模型: LPS (0.2 μg/mL) 处理的奶牛 [3] 剂量: 50 mg/kg 给药途径: 注射(每 30 分钟采集一次牛奶样本,直至 6 小时和 9 小时) 实验结果: 降低了牛奶中体细胞计数 (SCC)、血清白蛋白 (SA)、IgG 和乳酸脱氢酶 (LDH) 活性的增加。 1. 饮食诱导肥胖小鼠模型: - 动物:雄性 C57BL/6 小鼠(6 周龄,18-22 g),n=24,随机分为 4 组:正常饮食 (ND) 组、高脂饮食 (HFD) 组、HFD + 酮洛芬 10 mg/kg 组、HFD + 酮洛芬 30 mg/kg 组(每组 n=6)。 - 模型构建:ND组饲喂正常饮食(10%脂肪);HFD组饲喂高脂饮食(45%脂肪),持续8周以诱导肥胖。 - 给药:将酮洛芬(RP-19583)溶于0.5%二甲基亚砜(DMSO)+生理盐水(最终DMSO浓度≤0.1%)。从第9周到第12周,HFD+酮洛芬组每日灌胃给药(灌胃量:10 μL/g体重);ND组和HFD组灌胃给予溶剂(0.5% DMSO +生理盐水)。 - 样本采集:每周称量小鼠体重。第12周时,采用颈椎脱臼法处死小鼠;采集附睾白色脂肪组织(eWAT)、皮下白色脂肪组织(sWAT)、棕色脂肪组织(BAT)、肝脏和肾脏,称重后置于-80℃保存,以备后续分析[2]。 2. 奶牛乳腺炎症模型: - 动物:选取12头泌乳期荷斯坦奶牛(2-3胎,泌乳期150-200天),随机分为3组:对照组、LPS组和LPS+酮洛芬组(每组n=4)。 - 模型建立:LPS组和LPS+酮洛芬组在右后乳区进行LPS(100 μg/乳区)乳内注射;对照组进行生理盐水乳内注射。 - 给药:LPS+酮洛芬组在LPS注射后1小时进行酮洛芬(50 mg/乳区,溶于5 mL生理盐水)乳内注射;对照组和LPS组进行5 mL生理盐水乳内注射。 - 样本采集:分别于治疗后0、6、12和24小时进行乳腺组织活检;同时采集乳汁样本。于治疗后24小时经尾静脉采集血清[3] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
酮洛芬口服吸收迅速且良好,血浆峰浓度在0.5至2小时内达到。 24小时内,约80%的给药剂量经尿液排出,主要以葡萄糖醛酸苷代谢物的形式排出。 口服剂量清除率=6.9±0.8升/小时 [酮洛芬速释胶囊(4×50毫克)] 口服剂量清除率=6.8±1.8升/小时 [酮洛芬缓释胶囊(1×200毫克)] 0.08升/公斤/小时 0.7升/公斤/小时 [酒精性肝硬化患者] 代谢/代谢物 迅速且广泛地酮洛芬主要在肝脏代谢,主要通过与葡萄糖醛酸结合代谢。尚未发现活性代谢物。 酮洛芬已知的代谢物包括酮洛芬葡萄糖醛酸苷。 生物半衰期 普通胶囊:1.1-4 小时;缓释胶囊:5.4 小时(由于吸收延迟,其固有清除率与普通胶囊相同)。 |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
前瞻性研究表明,1%至2%服用酮洛芬的患者会出现至少短暂的血清转氨酶升高。即使继续用药,这些升高也可能自行消退。显著的转氨酶升高(升高3倍以上)发生于 可能性评分:C(可能是临床上明显的肝损伤的罕见原因)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 虽然酮洛芬在母乳中的浓度较低,但有一家中心报告称,他们收到过服用酮洛芬的母亲所生的母乳喂养婴儿出现肾脏和胃肠道不良反应的报告。最好选择其他药物,尤其是在哺乳新生儿或早产儿时。 ◉ 对母乳喂养婴儿的影响 法国药物警戒中心汇总了1985年1月至2011年6月期间在法国报告的所有母乳喂养婴儿不良反应。在174份报告中,酮洛芬被报道导致8名婴儿出现不良反应,并且是严重不良反应(例如食管溃疡、糜烂性胃炎、脑膜出血和肾功能不全)中最常被怀疑的药物之一。 ◉ 对哺乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白质结合 99%结合,主要与白蛋白结合 1. 小鼠毒性:在为期4周的饮食诱导肥胖研究中,10 mg/kg和30 mg/kg/天的酮洛芬(RP-19583)对小鼠肝脏重量/体重比(30 mg/kg组:3.2 ± 0.2% vs. 高脂饮食组:3.3 ± 0.2%)或肾脏重量/体重比(30 mg/kg组: 0.8 ± 0.1% vs. HFD 组:0.8 ± 0.1%。肝肾组织 HE 染色显示无病理变化(例如坏死、炎症)[2] 2. 奶牛毒性:在 24 小时乳腺炎症研究中,LPS + 酮洛芬组的血清丙氨酸氨基转移酶 (ALT) 水平 (45 ± 5 U/L) 与对照组 (43 ± 4 U/L) 相似;血清天冬氨酸氨基转移酶 (AST) 水平 (52 ± 6 U/L vs. 对照组 50 ± 5 U/L) 和肌酐水平 (0.8 ± 0.1 mg/dL vs. 对照组 0.7 ± 0.1 mg/dL) 也未显示显著差异,表明无肝毒性或肾毒性[3] |
| 参考文献 |
|
| 其他信息 |
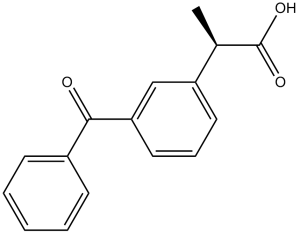
酮洛芬是一种氧代单羧酸,由丙酸在2位被3-苯甲酰苯基取代而成。它是一种非甾体抗炎药、解热药、EC 1.14.99.1(前列腺素内过氧化物合酶)抑制剂、环境污染物、外源性物质和药物过敏原。它属于二苯甲酮类化合物,是一种氧代单羧酸,在功能上与丙酸相关。
酮洛芬是丙酸衍生物,是一种具有镇痛和解热作用的非甾体抗炎药(NSAID)。 酮洛芬是一种非甾体抗炎药。酮洛芬的作用机制是作为环氧合酶抑制剂。 酮洛芬是一种非甾体抗炎药(NSAID),用于治疗急性疼痛和慢性关节炎。酮洛芬与治疗期间血清酶升高发生率较低以及罕见的临床表现明显的急性肝损伤病例相关。 已有关于酮洛芬在智人中应用的报道,并有相关数据。 酮洛芬是一种丙酸衍生物,属于非甾体抗炎药 (NSAID),具有抗炎、镇痛和解热作用。酮洛芬抑制环氧合酶 I 和 II 的活性,从而减少前列腺素和血栓素前体的生成。前列腺素合成酶介导的前列腺素合成减少,是酮洛芬发挥治疗作用的原因。酮洛芬还会减少血栓素合成酶介导的血栓素 A2 的生成,从而抑制血小板聚集。 一种布洛芬类抗炎镇痛解热药。它用于治疗类风湿性关节炎和骨关节炎。 另见:酮洛芬赖氨酸(其活性部分);酮洛芬钠(其活性部分);酮洛芬;土拉霉素(成分)……查看更多…… 药物适应症 用于治疗急性和慢性类风湿性关节炎、骨关节炎、强直性脊柱炎、原发性痛经以及与肌肉肌腱损伤(扭伤和拉伤)、术后(包括牙科手术)或产后疼痛相关的轻度至中度疼痛。 FDA标签 治疗肌肉骨骼和结缔组织疼痛 作用机制 酮洛芬的抗炎作用被认为是通过抑制环氧合酶-2 (COX-2) 实现的,COX-2 是一种参与花生四烯酸途径前列腺素合成的酶。这导致介导疼痛、发热和炎症的前列腺素水平降低。酮洛芬是一种非特异性环氧合酶抑制剂,抑制COX-1被认为是其部分副作用(例如胃肠道不适和溃疡)的根源。酮洛芬被认为具有抗缓激肽活性以及溶酶体膜稳定作用。其解热作用可能源于对下丘脑的作用,从而增加外周血流量、血管舒张并最终散热。 药效学 酮洛芬是一种具有镇痛和解热作用的非甾体抗炎药(NSAIDs)。酮洛芬的药理作用与其他典型的NSAIDs相似,均可抑制前列腺素的合成。酮洛芬用于治疗类风湿性关节炎、骨关节炎、痛经,并缓解中度疼痛。 1.酮洛芬(RP-19583)是一种非甾体抗炎药(NSAID),与母体类似物相比,其COX-2选择性有所提高(比值为2.9),这是通过基于结构的修饰增强与COX-2活性位点的结合而实现的[1] 2. 酮洛芬的抗肥胖作用是通过mTORC1-p38-COX-2信号通路介导的:mTORC1和p38的激活上调COX-2,进而促进白色脂肪褐变标志物(UCP1、PGC-1α)的表达,增加能量消耗并减少脂肪堆积[2] 3.在奶牛中,酮洛芬通过抑制 TLR4 介导的促炎细胞因子(IL-6、TNF-α)分泌并促进抗炎细胞因子(IL-10)的产生来减轻乳腺炎症,这在预防和治疗牛乳腺炎方面具有潜在的应用价值[3] |
| 分子式 |
C16H14O3
|
|
|---|---|---|
| 分子量 |
254.28
|
|
| 精确质量 |
254.094
|
|
| CAS号 |
22071-15-4
|
|
| 相关CAS号 |
Ketoprofen-d3;159490-55-8;Ketoprofen-d4;1219805-29-4;S-(+)-Ketoprofen;22161-81-5;Ketoprofen (lysinate);57469-78-0;Ketoprofen-13C,d3;1189508-77-7;Dexketoprofen (trometamol);156604-79-4
|
|
| PubChem CID |
3825
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.2±0.1 g/cm3
|
|
| 沸点 |
431.3±28.0 °C at 760 mmHg
|
|
| 熔点 |
94 °C
|
|
| 闪点 |
228.8±20.5 °C
|
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
|
| 折射率 |
1.592
|
|
| LogP |
2.81
|
|
| tPSA |
54.37
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
3
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
19
|
|
| 分子复杂度/Complexity |
331
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
CC(C1=CC(=CC=C1)C(=O)C2=CC=CC=C2)C(=O)O
|
|
| InChi Key |
DKYWVDODHFEZIM-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C16H14O3/c1-11(16(18)19)13-8-5-9-14(10-13)15(17)12-6-3-2-4-7-12/h2-11H,1H3,(H,18,19)
|
|
| 化学名 |
2-(3-benzoylphenyl)propanoic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (9.83 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (9.83 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (9.83 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.9327 mL | 19.6634 mL | 39.3267 mL | |
| 5 mM | 0.7865 mL | 3.9327 mL | 7.8653 mL | |
| 10 mM | 0.3933 mL | 1.9663 mL | 3.9327 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT02439034
Conditions:Uterine Cervical Cancer|Upper Aerodigestive Tract NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT06912763
Conditions:Fibrosis Syndrome|Lymphedema|Head &Amp; Neck Cancer|FibrosisLink: https://clinicaltrials.gov/ct2/show/NCT07319728
Conditions:Knee Osteoarthritis
Title:Efficacy of Oral Paracetamol Compared With Oral Ketoprofen for Pain Management in Office Hysteroscopy
Status:Completed
updateDate:2026-01-02
Ctid:NCT07315698
Link: https://clinicaltrials.gov/ct2/show/NCT07315698
Conditions:Office HysteroscopyLink: https://clinicaltrials.gov/ct2/show/NCT04726592
Conditions:Migraine|Migraine Without Aura|Migraine With Aura|HeadacheLink: https://clinicaltrials.gov/ct2/show/NCT06381063
Conditions:Acute Postoperative Pain|Non-steroidal Anti-inflammatory Drugs|Cardiac Surgery|Pain Intensity|Multimodal Pain ManagementLink: https://clinicaltrials.gov/ct2/show/NCT06731647
Conditions:Non-steroidal Anti-inflammatory Drugs|Pleurodesis|Pneumothorax|Pain ManagementLink: https://clinicaltrials.gov/ct2/show/NCT05062083
Conditions:Multiple SclerosisLink: https://clinicaltrials.gov/ct2/show/NCT02804126
Conditions:Cesarean Section|Pain Management|Neuropathic PainLink: https://clinicaltrials.gov/ct2/show/NCT03404908
Conditions:Cesarean Section|Postoperative PainLink: https://clinicaltrials.gov/ct2/show/NCT03783715
Conditions:LymphedemaLink: https://clinicaltrials.gov/ct2/show/NCT04272372
Conditions:Breast Cancer LymphedemaLink: https://clinicaltrials.gov/ct2/show/NCT02257970
Conditions:LymphedemaLink: https://clinicaltrials.gov/ct2/show/NCT04500470
Conditions:Infertility, FemaleLink: https://clinicaltrials.gov/ct2/show/NCT04421911
Conditions:OsteoarthritisLink: https://clinicaltrials.gov/ct2/show/NCT03244540
Conditions:Cesarean Section|Pain, Postoperative|Pain, NeuropathicLink: https://clinicaltrials.gov/ct2/show/NCT02400047
Conditions:Postoperative Nausea and VomitingLink: https://clinicaltrials.gov/ct2/show/NCT04148846
Conditions:Headache, Post-Dural PunctureLink: https://clinicaltrials.gov/ct2/show/NCT03585374
Conditions:Acute Traumatic PainLink: https://clinicaltrials.gov/ct2/show/NCT01882530
Conditions:Post Operative AnalgesiaLink: https://clinicaltrials.gov/ct2/show/NCT02905045
Conditions:Pain ReliefLink: https://clinicaltrials.gov/ct2/show/NCT02905058
Conditions:Pain ReliefLink: https://clinicaltrials.gov/ct2/show/NCT00188071
Conditions:Abortion, InducedLink: https://clinicaltrials.gov/ct2/show/NCT00557544
Conditions:MigraineLink: https://clinicaltrials.gov/ct2/show/NCT02638831
Conditions:Gonarthrosis|Low Back PainLink: https://clinicaltrials.gov/ct2/show/NCT02730026
Conditions:Pain|Other Surgical ProceduresLink: https://clinicaltrials.gov/ct2/show/NCT02248493
Conditions:Pain, Postoperative|Recovery of Function|ChildLink: https://clinicaltrials.gov/ct2/show/NCT02840526
Conditions:Postoperative PainLink: https://clinicaltrials.gov/ct2/show/NCT02099006
Conditions:Female Genital DiseasesLink: https://clinicaltrials.gov/ct2/show/NCT02538224
Conditions:Chronic PeriodontitisLink: https://clinicaltrials.gov/ct2/show/NCT01816334
Conditions:SciaticaLink: https://clinicaltrials.gov/ct2/show/NCT02491879
Conditions:Mechanical Low Back PainLink: https://clinicaltrials.gov/ct2/show/NCT02491736
Conditions:Ankle SprainLink: https://clinicaltrials.gov/ct2/show/NCT02393846
Conditions:Ankle SprainLink: https://clinicaltrials.gov/ct2/show/NCT00799838
Conditions:PharyngotonsillitisLink: https://clinicaltrials.gov/ct2/show/NCT02238340
Conditions:SurgeryLink: https://clinicaltrials.gov/ct2/show/NCT02180087
Conditions:Cardiac Surgery|Post-operative PainLink: https://clinicaltrials.gov/ct2/show/NCT01890902
Conditions:Acute Pain (Flare) Associated With Osteoarthritis (OA) of the KneeLink: https://clinicaltrials.gov/ct2/show/NCT00765700
Conditions:Sprain|Strain|Acute Soft Tissue InjuryLink: https://clinicaltrials.gov/ct2/show/NCT00553605
Conditions:PainLink: https://clinicaltrials.gov/ct2/show/NCT01223053
Conditions:SprainLink: https://clinicaltrials.gov/ct2/show/NCT01373697
Conditions:Muscular Atrophy|Sprains|TendonitisLink: https://clinicaltrials.gov/ct2/show/NCT01020279
Conditions:Musculoskeletal PainLink: https://clinicaltrials.gov/ct2/show/NCT00679146
Conditions:Low Back PainLink: https://clinicaltrials.gov/ct2/show/NCT00929877
Conditions:PainLink: https://clinicaltrials.gov/ct2/show/NCT00810121
Conditions:PainLink: https://clinicaltrials.gov/ct2/show/NCT00372333
Conditions:Joint Pain|Musculoskeletal Pain|Stiffness|Soft Tissue Inflammation in Designated Target Area(s)Link: https://clinicaltrials.gov/ct2/show/NCT00211549
Conditions:Osteoarthritis, KneeLink: https://clinicaltrials.gov/ct2/show/NCT00316784
Conditions:Osteoarthritis; KneeLink: https://clinicaltrials.gov/ct2/show/NCT00265304
Conditions:Osteoarthritis, KneeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-023888-17
Condition:Arthroscopic surgeryLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-021240-18
Condition:Elective Laparoscopic colectomyIntervento di colectomia laparoscopica in elezioneLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-021851-23
Condition:Inguinal hernia elective surgeryIntervento di ernioplastica inguinale in elezioneLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-013864-37
Condition:acute soft tissue injury minor traumatism (such as: sprain, strain, dislocation,...etc.) or contusionLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-004484-18
Condition:Tutkimukseen otettavat potilaat ovat perusterveitä, primääriin lonkan tekonivelleikkaukseen tulevia henkilöitäLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2006-006298-26
Condition:Treatment of osteoarthritis of the kneeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-002086-31
Condition:Acute Migraine in childhoodLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-005388-85
Condition:ANALGESIE POST OPERATOIRE après anesthésie locorégionale intraveineuseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-002581-35
Condition:The study population is a healthy population who are voluntarily participating in this clinical trial. This study will compare the PK profile after e.c. and oral application of the same dosage of ketoprofen and in a dosage range as intended for clinical use in OA. It will also indicate whether application on muscle will result in a different PK profile as compared to application on a joint.The intended use for Diractin® will be osteoarthritis of the kneeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2006-000383-88
Condition:Painful, acute ankle sprains, as a model of general traumatic soft-tissue injuriesLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2004-001239-42
Condition:treatment of bruised or sprained jointsLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-002250-21
Condition:ANTALGIC EFFECT AND TOLERABILITY OF A NEW DOSAGE OF PARACETAMOL SYRUP IN PHARINGOTONSILLYTIS IN PEDIATRYLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2004-002398-22
Condition:Patients with different dermatological diseases· Atopic eczema (MedDRA 6.0, LLT: 10003641)· Dishydrotic hand eczema (MedDRA 6.0, LLT: 10013913)· Plaque type psoriasis (MedDRA 6.0, LLT: 10050576)· Seborrheic eczema (MedDRA 6.0, LLT: 10039795)· Acne vulgaris (MedDRA 6.0, LLT: 10000519)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2004-002856-32
Condition:non articular rheumatismLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2004-000843-61
Condition:ANKLE SPRAINLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-002875-34
Condition:Patients suffering from actinic keratosis to be treated with PDTLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2014-005026-35
Condition:Per-operating analgesia, pediatric ambulatory surgeryAnalgésie peropératoire, chirurgie ambulatoire pédiatriqueInvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
