| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
- Mavoglurant (AFQ056) selectively targets metabotropic glutamate receptor 5 (mGluR5) as a negative allosteric modulator; the Ki value was reported as 14 nM in radioligand binding assays [1]
- No significant binding affinity was observed for other mGluR subtypes (mGluR1/2/3/4/6/7/8) at concentrations up to 10 μM [1] |
|---|---|
| 体外研究 (In Vitro) |
- 在表达人mGluR5的HEK293细胞中,马沃格鲁兰(Mavoglurant,AFQ056) 抑制quisqualate诱导的钙动员,IC50为33 nM [1]
- 在大鼠皮质神经元中呈剂量依赖性减少谷氨酸诱导的肌醇磷酸积累,1 μM时达到最大抑制率(72%) [1] - 在脆性X综合征患者的原代成纤维细胞中,马沃格鲁兰(Mavoglurant,AFQ056)(1 μM)可逆转由FMR1基因沉默引起的蛋白质合成亢进 [2] Mavoglurant(1 nM-10 μM;10 分钟)完全拮抗 hmGluR5 介导的反应,在稳定表达 mGluR5a 的 L(tk-) 细胞中的 Ca2+- 和 PI 周转测试中,IC50 分别为 110 和 30 nM [1]。 Mavoglurant (0.01 nM-10 μM) 在大鼠脑膜中以浓度依赖性方式取代变构结合配体 [3H]-AAE327 的结合,IC50 为 47 nM [1]。 |
| 体内研究 (In Vivo) |
- 在Fmr1基因敲除小鼠(脆性X综合征模型)中,口服马沃格鲁兰(Mavoglurant,AFQ056)(10 mg/kg)显著改善听源性癫痫(减少70%)和多动行为(运动活性降低35%) [1]
- 在剂量≥3 mg/kg时恢复Fmr1敲除小鼠的海马长时程抑制(LTD),通过场兴奋性突触后电位(fEPSP)记录证实 [1] - 在6-OHDA损伤的帕金森病大鼠模型中,马沃格鲁兰(Mavoglurant,AFQ056)(3 mg/kg,腹腔注射)减少45%的左旋多巴诱导异动症,且不影响抗帕金森疗效 [3] mavoglurant(0.1-10 mg/kg;单次口服剂量)以剂量依赖性方式抑制小鼠应激性高热(SIH)[1]。 Mavoglurant 单次口服剂量为 9.4 mg/kg,终末半衰期为 2.9 小时,中等口服生物利用度为 32%,脑和血浆中的 Cmax 分别为 950 pmol/mL 和 3500 pmol/g。分别[1]。 Mavoglurant(3.1 mg/kg;静脉注射;单剂量)的 Tmax 小于 0.08 小时,Cmax(血浆;脑)为 3330 pmol/mL,终末半衰期为 0.69 小时 [1]。 |
| 酶活实验 |
- 放射性配体结合实验:
- 将mGluR5表达细胞的膜制剂与[³H]MPEP(选择性mGluR5配体)及系列稀释的马沃格鲁兰(Mavoglurant,AFQ056)(0.01-1000 nM)在结合缓冲液(pH 7.4)中25°C孵育90分钟。
- 过滤分离结合态配体,通过放射性测量计算置换效力和Ki值 [1]
- 钙动员实验: - 向负载钙敏感染料的mGluR5转染HEK293细胞中加入quisqualate(1 μM)和马沃格鲁兰(Mavoglurant,AFQ056)。 - 监测300秒内的荧光强度变化,确定受体介导的钙反应抑制IC50 [1] |
| 细胞实验 |
- 脆性X成纤维细胞蛋白质合成实验:
- 来自FMR1全突变患者的原代成纤维细胞用马沃格鲁兰(Mavoglurant,AFQ056)(0.1-10 μM)处理24小时。
- 通过[³H]亮氨酸掺入法测量蛋白质合成,结果归一化至总蛋白含量 [2]
- 肌醇磷酸积累实验: - 大鼠皮质神经元用马沃格鲁兰(Mavoglurant,AFQ056)预孵育30分钟,随后用谷氨酸(100 μM)刺激1小时。 - 提取积累的肌醇磷酸并通过闪烁计数定量 [1] |
| 动物实验 |
动物/疾病模型: 雄性 OF1/IC 小鼠 [1]
剂量: 0.1、1、10 mg/kg 给药途径: 单次口服 实验结果: 降低应激诱导的高热。其效果与阳性对照氯氮卓相当。 动物/疾病模型: 雄性 SD(SD(Sprague-Dawley))大鼠(175-250 g)[1] 剂量: 3.1 mg/kg 静脉注射 (iv);9.4 mg/kg 口服(药代动力学/PK/PK 分析) 给药途径: 单次静脉注射或口服 实验结果: 口服:F=32%; T1/2=2.9 小时;Tmax≤0.25 小时。静脉注射:T1/2=0.69 小时;Cmax(血浆/脑组织)=3330 pmol·mL-1/8400 pmol·g-1;Tmax≤0.08 小时。 - 脆性 X 综合征小鼠模型:- 雄性 Fmr1 基因敲除小鼠(8-12 周龄)每日一次口服 Mavoglurant (AFQ056)(溶于 0.5% 甲基纤维素),剂量分别为 1、3 或 10 mg/kg,连续 7 天。 - 在末次给药后 1 小时进行行为学测试(旷场实验、听源性癫痫易感性测试),随后采集海马组织进行电生理分析[1] - 帕金森病大鼠模型: - 单侧 6-OHDA 损伤的大鼠在给予左旋多巴 (6 mg/kg) 前 30 分钟腹腔注射 Mavoglurant (AFQ056) (1 或 3 mg/kg),每日一次,持续 21 天。 - 在治疗期间,使用异常不自主运动 (AIM) 量表对运动障碍严重程度进行评分[3] |
| 药代性质 (ADME/PK) |
小鼠口服Mavoglurant (AFQ056)(10 mg/kg)后,血浆峰浓度 (Cmax) 在 1.2 小时达到,生物利用度为 65% [1]
- 该化合物在大鼠中显示出良好的脑渗透性,脑血浆比为 2.3 [1] - 在啮齿动物中,血浆消除半衰期约为 4.5 小时 [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
- 在为期 28 天的大鼠重复给药毒性研究中,剂量高达 30 mg/kg/天的 Mavoglurant (AFQ056) 未引起体重、血液学参数或肝肾功能指标的显著变化 [1]
- 在标准体外试验中未观察到致突变性或遗传毒性 [1] - 在人和大鼠血浆中的血浆蛋白结合率均 >99% [1] |
| 参考文献 |
|
| 其他信息 |
Mavoglurant (AFQ056) 是通过吡啶衍生物的构效关系 (SAR) 优化发现的,与早期的 mGluR5 拮抗剂相比,其代谢稳定性有所提高 [1]
- 在脆性 X 综合征患者中,对 Mavoglurant (AFQ056) 的反应与 FMR1 基因甲基化状态相关,甲基化水平 <5% 的患者表现出更好的行为改善 [2] - 帕金森病的临床开发重点在于减少左旋多巴引起的运动障碍,II 期试验显示,每日 100-200 mg 的剂量具有显著疗效 [3] Mavoglurant 已用于研究对 SSRI 治疗无效的强迫症患者(在适当剂量下使用 SSRI 治疗 12 周无效)的治疗试验。 |
| 分子式 |
C19H23NO3
|
|---|---|
| 分子量 |
313.397
|
| 精确质量 |
313.167
|
| CAS号 |
543906-09-8
|
| 相关CAS号 |
Mavoglurant racemate;1636881-61-2
|
| PubChem CID |
9926832
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
476.3±45.0 °C at 760 mmHg
|
| 闪点 |
241.8±28.7 °C
|
| 蒸汽压 |
0.0±1.3 mmHg at 25°C
|
| 折射率 |
1.602
|
| LogP |
3.51
|
| tPSA |
49.77
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
519
|
| 定义原子立体中心数目 |
3
|
| SMILES |
CC1=CC(=CC=C1)C#C[C@@]2(CCC[C@@H]3[C@H]2CCN3C(=O)OC)O
|
| InChi Key |
ZFPZEYHRWGMJCV-ZHALLVOQSA-N
|
| InChi Code |
InChI=1S/C19H23NO3/c1-14-5-3-6-15(13-14)8-11-19(22)10-4-7-17-16(19)9-12-20(17)18(21)23-2/h3,5-6,13,16-17,22H,4,7,9-10,12H2,1-2H3/t16-,17-,19-/m1/s1
|
| 化学名 |
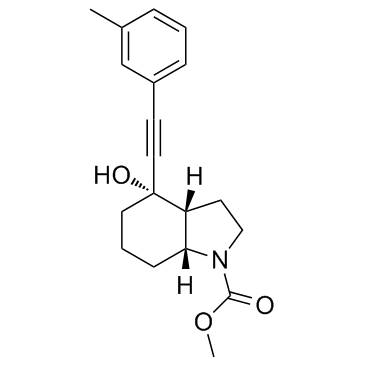
methyl (3aR,4S,7aR)-4-hydroxy-4-[2-(3-methylphenyl)ethynyl]-3,3a,5,6,7,7a-hexahydro-2H-indole-1-carboxylate
|
| 别名 |
AFQ 056; AFQ-056; AFQ056
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~120 mg/mL (~382.91 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 3 mg/mL (9.57 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 30.0 mg/mL 澄清的 DMSO 储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 3 mg/mL (9.57 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 30.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 3 mg/mL (9.57 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1908 mL | 15.9541 mL | 31.9081 mL | |
| 5 mM | 0.6382 mL | 3.1908 mL | 6.3816 mL | |
| 10 mM | 0.3191 mL | 1.5954 mL | 3.1908 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT05203965
Conditions:Familial Alcoholism VulnerabilityLink: https://clinicaltrials.gov/ct2/show/NCT06136195
Conditions:Alcohol Consumption|Heavy Drinker|Alcohol Use Disorder (AUD)Link: https://clinicaltrials.gov/ct2/show/NCT03327792
Conditions:Alcohol Drinking
Title:AFQ056 for Language Learning in Children With FXS
Status:Completed
updateDate:2023-10-10
Ctid:NCT02920892
Link: https://clinicaltrials.gov/ct2/show/NCT02920892
Conditions:Fragile X SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT03341715
Conditions:Familial Alcoholism VulnerabilityLink: https://clinicaltrials.gov/ct2/show/NCT03242928
Conditions:Cocaine-related DisorderLink: https://clinicaltrials.gov/ct2/show/NCT04771143
Conditions:Cocaine Use DisorderLink: https://clinicaltrials.gov/ct2/show/NCT01173731
Conditions:Parkinson Disease|Dyskinesia, Drug-Induced|LevodopaLink: https://clinicaltrials.gov/ct2/show/NCT01491529
Conditions:Dyskinesias|Parkinson Disease|Movement Disorders|Parkinsonian Disorders|Anti-Dyskinesia AgentsLink: https://clinicaltrials.gov/ct2/show/NCT01385592
Conditions:Dyskinesias|Parkinson Disease|Movement Disorders|Parkinsonian DisordersLink: https://clinicaltrials.gov/ct2/show/NCT01491932
Conditions:Dyskinesias|Parkinson Disease|Movement Disorders|Parkinsonian Disorders|Anti-Dyskinesia AgentsLink: https://clinicaltrials.gov/ct2/show/NCT01253629
Conditions:Fragile X SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT01442259
Conditions:Mild Moderate|or Severe Renal ImpairmentLink: https://clinicaltrials.gov/ct2/show/NCT01456663
Conditions:Hepatic ImpairmentLink: https://clinicaltrials.gov/ct2/show/NCT01482143
Conditions:Fragile X SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT00986414
Conditions:Parkinson Disease|DyskinesiasLink: https://clinicaltrials.gov/ct2/show/NCT01813019
Conditions:Patient Diagnosed With OCD and Resistant to SSRI Treatment|Failed SSRI Over 12 Weeks at Appropriate DosesLink: https://clinicaltrials.gov/ct2/show/NCT01348087
Conditions:Fragile X SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT00888004
Conditions:Parkinson's Disease|L-dopa Induced DyskinesiaLink: https://clinicaltrials.gov/ct2/show/NCT01433354
Conditions:Fragile X SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT01357239
Conditions:Fragile X SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT01019473
Conditions:Huntington's Disease|ChoreaLink: https://clinicaltrials.gov/ct2/show/NCT00582673
Conditions:Parkinson's DiseaseLink: https://clinicaltrials.gov/ct2/show/NCT00414856
Conditions:Gastroesophageal Reflux DiseaseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-002379-40
Condition:Fragile X syndromeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-002073-30
Condition:L-dopa induced dyskinesias in patients with Parkinson?s diseaseDiscinesias inducidas por levodopa en pacientes con enfermedad de ParkinsonLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-001952-12
Condition:Fragile X SyndromeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-022638-96
Condition:Fragile X SyndromeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-000365-12
Condition:Not Applicable. This is a PK study (including evaluation of food effect).Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-013667-19
Condition:Fragile X SyndromeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-019418-25
Condition:pazienti conmalattia di Parkinson con discinesie indotte dalla L-dopa.Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-011743-39
Condition:Huntington's diseaseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-008712-98
Condition:moderate to severe levodopa induced dyskinesias in patients with Parkinson's diseaseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-006270-15
Condition:L-dopa induced dyskinesias in Parkinson's patients with severe motor complicationsLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-005088-82
Condition:Fragile X SyndromeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-002900-16
Condition:The main objective of this study is to assess the efficacy of AFQ056 in reducing L-dopa induced dyskinesias.InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
