| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
Catechol-O-methyltransferase (COMT),ED50 < 1.4 mg⋅kg(-1)
|
|---|---|
| 体外研究 (In Vitro) |
阿匹卡彭可延长 L-DOPA 的生物利用度,且不会造成伤害,因为它会长时间抑制外周 COMT。 Opicapone 的 IC50 值为 98 μM,表明细胞 ATP 水平降低。将人原代肝细胞置于浓度不断增加的 Ro 40-7592、OR-611 或 Opicapone 一整天后,JC-1 聚集体与 JC-1 的比率 评估单体(λex 544 λem 590 与 λex 485 λem 的比率) 538)揭示了细胞线粒体膜电位的浓度依赖性降低。 Opicapone 的 IC50 为 181 μM,可降低细胞线粒体膜的电位[1]。
|
| 体内研究 (In Vivo) |
opticapartone 可抑制大鼠的外周 COMT,注射后 6 小时其 ED50 值低于 1.4 mg/kg。在最初的 8 小时内,效果仍然存在,到 24 小时后,COMT 尚未恢复到控制水平。虽然 Ro 40-7592 在给药后仅两小时就显示出显着效果,但单次注射 Opicapone 导致血浆 L-DOPA 水平持续升高,并在给药后 2 至 24 小时内同时降低 3-OMD。服用 L-DOPA 后,阿片卡朋对大脑儿茶酚胺的影响可持续长达 24 小时。经过 24 小时潜伏期后,阿片卡彭也是降低人原代肝细胞 ATP 水平和线粒体膜电位效率最低的药物 [1]。
|
| 酶活实验 |
COMT活性
在实验当天,将红细胞在冰中解冻,并通过添加四体积的MilliQ水进行溶血。剧烈混合后,将试管在冰上保持10 分钟,然后在20 000× g在4°C下持续20 分钟。收集的上清液用于COMT活性测定。组织在冰上解冻。肝碎片在Precellys 24双组织匀浆器(Bertin Corporation,Washington DC,USA)中匀浆两个循环,每次5 s,间隔5 冰上分钟。肾脏和大脑用Silent Crusher M匀浆器(Heidolph,Schwabach,Germany)和探针8F/M匀浆约45 s处于最大速度。匀浆用于COMT活性测定。通过BioRad蛋白质测定法(BioRad,Hercules,CA,USA)使用BSA的标准曲线(50–250)测定匀浆和溶血样品中的总蛋白质 μg·mL−1)。[1] 使用肾上腺素作为底物测定COMT活性,并测量如前所述形成的后肾上腺素(Bonifácio et al 等人,2003年)。简而言之,反应混合物(总体积为1000 μL)含有500 μL样品(2 mg总蛋白),帕吉林(100 μM),氯化镁(100 μM),EGTA(1 mM)、S-腺苷甲硫氨酸(500 μM用于肝脏和红细胞;250 μM用于肾脏和100 μM用于大脑)和肾上腺素(1000 μM用于肝脏、红细胞、肾脏和100 μM用于大脑)在5 mM磷酸盐缓冲液pH 7.8. 用底物开始反应,然后进行5 分钟(肝脏),10 min(红细胞和肾脏)或15 37°C时的min(脑)。通过添加200停止反应 μL 2 M PCA,脱蛋白后,将样品注射到HPLC-ED上。[1] |
| 细胞实验 |
ATP测定
使用ATP-Lite测定系统(Perkin-Elmer,Waltham,MA,USA)测定人原代肝细胞的ATP含量,该系统基于ATP与添加的荧光素酶和D-荧光素反应产生的光。接种24小时后,用Hank平衡盐溶液(HBSS)洗涤细胞培养物,然后与在无胎牛血清的培养基中制备的测试化合物(0、1.56、3.13、6.25、12.5、25、50、100和200 μM)24 在37°C、湿度为5%CO2-95%的空气中加热h。阳性对照(与羰基氰化物-三氟甲氧基苯基腙-FCCP孵育的细胞,10和50 μM)并行运行。孵育后,从孔中取出培养基并用100 μL HBSS加50 μL细胞裂解液。将盘子摇晃5分钟 最小400 室温下的r.p.m。基质溶液(50 μL)加入到每个孔中,并再次摇动平板5 最小400 r.p.m.,室温,光线柔和。三种标准浓度的ATP(1、10和100 μM)和空白在没有细胞的平板孔中平行运行。盘子是深色的,适合10个 min,并在MicrobetaTriLux(Perkin-Elmer)闪烁计数器上测定发光。[1] 线粒体膜电位测定 使用荧光染料JC-1对人原代肝细胞的线粒体膜电位进行评估。JC-1选择性地进入线粒体,而在具有高线粒体膜电位的健康细胞中,JC-1自发地形成具有强烈红色荧光的复合物,在具有低膜电位的凋亡或不健康细胞中JC-1保持显示绿色荧光的单体形式。接种24小时后,用HBSS和100洗涤细胞培养物 μL 15 μM的JC-1。1之后 h在37°C的黑暗中孵育,从孔中取出溶液,并用100洗涤细胞一次 μL HBSS。然后将细胞与在不含FBS的培养基中制备的测试化合物(1.56、3.13、6.25、12.5、25、50、100和200 μM)24 在37°C、湿度为5%CO2-95%的空气中加热h。阴性对照(无化合物)和阳性对照(用FCCP孵育的细胞,10和50 μM)并行运行。孵育后,从细胞中取出培养基并用100 μL HBSS。使用荧光微孔板读取器(Spectramax Gemini,Molecular Devices,Sunnyvale,CA,USA)在λex485下测量荧光 nm,λem538 nm(绿色)和λex544 nm,λem590 nm(红色)。线粒体膜电位计算为:比值=λex544λem590/λex485λem538。[1] |
| 动物实验 |
本研究使用了240只雄性Wistar大鼠。在评估该化合物对COMT抑制作用的实验中,动物分别给予不同剂量的奥匹卡朋(0.03、0.1、0.3、0.6、1、3和10 mg/kg),并在给药后2小时和6小时处死。在评估COMT时间-活性曲线的实验中,动物分别给予奥匹卡朋(3 mg/kg),并在给药后不同时间点(15分钟、30分钟、1小时、2小时、4小时、8小时、18小时、24小时和48小时)处死。在旨在评估化合物对中枢儿茶酚胺的影响的实验中,给动物注射了 3 mg/kg 的奥匹卡朋或 Ro 40-7592,并在安乐死前一小时给动物注射了 L-DOPA/苯甲氧基苯(L-DOPA 12 mg/kg 和苯甲氧基苯 3 mg/kg)。[1]
|
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服奥匹卡朋的吸收呈线性剂量依赖性。奥匹卡朋吸收迅速,口服生物利用度约为20%。单次服用50 mg奥匹卡朋后,中位达峰时间(Tmax)为2小时,范围为1至4小时。中等脂肪或中等热量的膳食可使Cmax降低62%,平均总血浆暴露量(AUC)降低31%,Tmax降低4小时。 健康受试者单次服用100 mg放射性标记的奥匹卡朋后,约70%的总剂量从粪便中回收,其中22%以原药形式排出。约20%的总剂量从呼出气体中回收,约5%从尿液中回收,其中不到1%的回收剂量以原药形式排出。尿液中检测到的主要代谢物是葡萄糖醛酸苷代谢物。 口服给药后,50 mg 奥匹卡朋的表观分布容积 (Vd) 为 29 L,个体间变异系数为 36%。一项研究显示,多次给药后有少量全身蓄积。 口服 50 mg 奥匹卡朋后,表观全身清除率为 22 L/h,个体间变异系数为 45%。 代谢/代谢物 根据临床和体外研究,硫酸化是奥匹卡朋的主要代谢途径,生成无活性代谢物。奥匹卡朋还可以进行葡萄糖醛酸化、COMT 介导的甲基化、还原和谷胱甘肽结合。作为两种主要的循环代谢物,BIA 9-1103(3-O-硫酸化奥匹卡朋)占总放射性的67.1%,BIA 9-1104(4-O-甲基化奥匹卡朋)占总放射性的20.5%。其他代谢物通常无法在血浆样本中进行定量分析。奥匹卡朋可发生N-氧化物还原反应生成BIA 9-1079,非临床研究表明BIA 9-1079是一种活性代谢物;然而,在人体内通常无法检测到BIA 9-1079。其他非活性代谢物包括BIA 9-1100、BIA 9-1101和BIA 9-1106。 生物半衰期 奥匹卡朋的平均消除半衰期为1至2小时。尽管半衰期很短,但观察到的奥匹卡朋诱导的 COMT 抑制在人类红细胞中的半衰期为 61.6 小时,标准差为 37.6 小时。 |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
在上市前对照试验中,奥匹卡朋治疗组患者血清ALT升高并不常见,且发生率与安慰剂对照组相似。在超过1000例接受奥匹卡朋治疗的患者的研究中,未发生严重肝脏事件,血清酶也未出现相关变化。奥匹卡朋获批上市并广泛应用后,尚未有临床上明显的肝损伤病例报告。然而,其临床应用经验有限。 可能性评分:E(不太可能是临床上明显的肝损伤的原因)。 蛋白结合 奥匹卡朋与血浆蛋白的结合率>99%,且与药物浓度无关。 |
| 参考文献 |
[1]. Pharmacological profile of Opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015 Apr;172(7):1739-52.
[2]. Opicapone as an adjunct to L-DOPA in patients with Parkinson's disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016 Feb;15(2):154-165 |
| 其他信息 |
奥匹卡朋是一种环状化合物,属于恶二唑类。
奥匹卡朋是一种强效、可逆的、外周作用的第三代儿茶酚-O-甲基转移酶 (COMT) 抑制剂。COMT 是一种参与多种儿茶酚胺(包括多巴胺)分解的酶。许多接受左旋多巴联合多巴脱羧酶 (DDC) 抑制剂(例如卡比多巴)治疗的帕金森病患者,随着时间的推移会出现运动并发症,因此需要使用多巴胺激动剂、单胺氧化酶 B 抑制剂(司来吉兰、雷沙吉兰)、儿茶酚-O-甲基转移酶 (COMT) 抑制剂或金刚烷胺,或使用缓释左旋多巴制剂来控制这些症状。奥匹卡朋用于治疗成人帕金森病患者,作为左旋多巴和卡比多巴的辅助治疗药物,尤其适用于治疗剂量末期运动波动。奥匹卡朋于2016年6月获得欧盟委员会批准,并于2020年4月获得美国食品药品监督管理局(FDA)批准上市。它以商品名Ongentys销售,为每日一次口服胶囊。奥匹卡朋作用持续时间超过24小时,可每日一次给药,与其他儿茶酚-O-甲基转移酶抑制剂相比,其细胞毒性风险最低。 奥匹卡朋是一种儿茶酚-O-甲基转移酶抑制剂。其作用机制是抑制儿茶酚-O-甲基转移酶。 奥匹卡朋是一种儿茶酚-O-甲基转移酶抑制剂,可作为左旋多巴/卡比多巴的辅助治疗药物,用于治疗帕金森病患者,尤其适用于治疗期间出现“关期”(运动症状复发)的情况。奥匹卡朋治疗期间血清酶升高发生率极低,且未发现与临床上明显的肝损伤伴黄疸病例相关。 药物适应症 奥匹卡朋适用于帕金森病成人患者,这些患者在服用左旋多巴和多巴脱羧酶抑制剂(例如卡比多巴)联合治疗后,症状仍无法稳定,且存在剂量末期运动波动或“关期”症状。 安替利夫适用于帕金森病成人患者,这些患者在服用左旋多巴/多巴脱羧酶抑制剂(DDCI)联合治疗后,症状仍无法稳定,且存在剂量末期运动波动。 昂格蒂斯适用于帕金森病成人患者,这些患者在服用左旋多巴/多巴脱羧酶抑制剂(DDCI)联合治疗后,症状仍无法稳定,且存在剂量末期运动波动。在这些组合疗法中病情稳定。Ongentys适用于帕金森病成人患者,作为左旋多巴/多巴脱羧酶抑制剂(DDCI)制剂的辅助治疗,这些患者在服用这些组合疗法后仍会出现剂量末期运动波动,且无法稳定病情。 作用机制 左旋多巴(L-Dopa)是治疗帕金森病运动症状和部分非运动症状的金标准;然而,只有一小部分给药的左旋多巴能够穿过血脑屏障发挥其治疗作用,患者面临着出现剂量末期运动波动的风险,这反映了左旋多巴在外周被芳香族L-氨基酸脱羧酶和儿茶酚-O-甲基转移酶(COMT)快速代谢。奥匹卡朋是一种外周选择性可逆性儿茶酚-O-甲基转移酶(COMT)抑制剂。它具有亚皮摩尔级的高结合亲和力,导致复合物解离速率常数缓慢,体内作用持续时间长。当奥匹卡朋加入含有左旋多巴和多巴脱羧酶抑制剂的治疗方案中时,奥匹卡朋有助于提高左旋多巴的血浆浓度,增强其治疗效果。 |
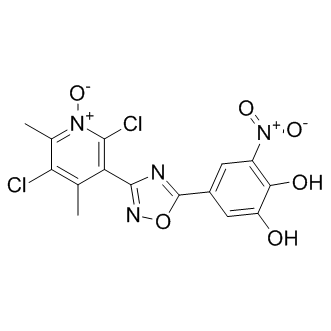
| 分子式 |
C15H10CL2N4O6
|
|---|---|
| 分子量 |
413.167
|
| 精确质量 |
411.998
|
| 元素分析 |
C, 43.61; H, 2.44; Cl, 17.16; N, 13.56; O, 23.23
|
| CAS号 |
923287-50-7
|
| PubChem CID |
135565903
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.80±0.1 g/cm3 (20 °C, 760 mmHg)
|
| 沸点 |
701.1±70.0 °C (760 mmHg)
|
| LogP |
4.598
|
| tPSA |
150.66
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
557
|
| 定义原子立体中心数目 |
0
|
| SMILES |
[O-][N+](C1C(O)=C(O)C=C(C2ON=C(C3C(C)=C(Cl)C(C)=[N+]([O-])C=3Cl)N=2)C=1)=O
|
| InChi Key |
ASOADIZOVZTJSR-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C15H10Cl2N4O6/c1-5-10(13(17)20(24)6(2)11(5)16)14-18-15(27-19-14)7-3-8(21(25)26)12(23)9(22)4-7/h3-4,22-23H,1-2H3
|
| 化学名 |
2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine 1-oxide
|
| 别名 |
BIA-91067; BIA 91067; BIA 9-1067
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~242.03 mM)
H2O : < 0.1 mg/mL |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.05 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (6.05 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.1 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + + 45% Saline 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4203 mL | 12.1016 mL | 24.2031 mL | |
| 5 mM | 0.4841 mL | 2.4203 mL | 4.8406 mL | |
| 10 mM | 0.2420 mL | 1.2102 mL | 2.4203 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
