| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
Estrogen receptor [DC50 = 2 nM]; CRBN
Vepdegestrant targets the estrogen receptor alpha (ERα), a key driver of cell growth in ER+ breast cancer . Its mechanism of action involves simultaneously binding to ER and the E3 ubiquitin ligase component cereblon (CRBN) . This interaction forms a ternary complex, leading to the ubiquitination of ER and its subsequent recognition and degradation by the proteasome. It is highly effective against both wild-type and clinically relevant, endocrine therapy-resistant ER mutants, such as ESR1 Y537S and D538G . |
|---|---|
| 体外研究 (In Vitro) |
ARV-471是一种雌激素受体(ER)α-PROTAC,是一种杂双功能分子,促进雌激素受体α与细胞内E3连接酶复合物之间的相互作用,导致雌激素受体通过蛋白酶体泛素化并随后降解。ARV-471以~2nM的半最大降解浓度(DC50)强力降解ER-阳性乳腺癌症细胞系中的ER。PROTAC介导的ER降解降低了经典调节的ER靶基因(PR、GREB1、TFF)的表达,并抑制了ER依赖性细胞系(MCF7、T47D)的细胞增殖。此外,ARV-471可降解临床相关的ESR1变体(Y537S和D538G),并抑制表达这些变体的细胞系的生长[2]。
在体外实验中,vepdegestrant可在多种ER+乳腺癌细胞系中强效、快速地降解ER。其半数降解浓度(DC50)约为1 nM,最大降解率(Dmax)超过95%。在100 nM浓度下,vepdegestrant在4小时内即可将ER蛋白水平降低80%以上。它对临床相关的、非配体依赖性ER突变体(如Y537S, D538G)同样有效,并能有效抑制ER依赖性乳腺癌细胞的增殖。 |
| 体内研究 (In Vivo) |
在未成熟的大鼠子宫营养模型中,ARV-471会降解大鼠子宫ER,并且没有表现出激动剂活性。每日口服单药ARV-471(3、10和30 mpk)会导致雌二醇依赖性MCF7异种移植物的肿瘤体积显著缩小,并在研究结束时伴随肿瘤ER蛋白减少>90%。此外,当CDK4/6抑制剂在MCF7模型中与ARV-471联合使用时,观察到更明显的肿瘤生长抑制(约130%TGI),同时ER蛋白水平显著降低。在ESR1 Y537S,激素非依赖性患者来源的异种移植物模型中,10 mpk的ARV-471完全抑制了生长,也降低了突变ER蛋白水平。综上所述,ARV-471的临床前数据支持其作为同类最佳口服ER PROTAC降解剂的持续发展[2]。
Vepdegestrant在MCF7原位异种移植物模型中实现了显著的TGI(87%-123%),优于ET药物氟维司群(31%-80%TGI)。在激素非依赖性(HI)突变ER Y537S患者源性异种移植物(PDX)癌症模型ST941/HI中,vepdegestrant实现了肿瘤消退,并且在ST941/HI/PBR帕博昔单抗耐药性模型(102%TGI)中同样有效。Vepdegestrant与CDK4/6抑制剂palbociclib、abemaciclib和ribociclib中的每一种联合使用可诱导稳健的肿瘤消退;mTOR抑制剂依维莫司;PI3K抑制剂alpelisib和inavolisib。 结论:与富司琼相比,维替格斯特在体内实现了更大的ER降解,这与TGI的改善有关,表明维替格斯特朗可能是治疗ER+/HER2-乳腺癌症患者更有效的骨干ET。 |
| 酶活实验 |
用于确认Vepdegestrant直接结合雌激素受体的主要非细胞学方法是无细胞放射性配体置换实验。在此流程中,首先将重组雌激素受体蛋白与高亲和力的放射性标记配体(如雌二醇)一同孵育。随后,向反应混合物中加入梯度浓度的Vepdegestrant。在充分孵育以达到结合平衡后,使用适当方法(通常为葡聚糖包被炭末法或过滤法)将结合态的配体与游离配体分离。最后,通过闪烁计数器检测仍与ER结合的放射性标记配体的量。Vepdegestrant竞争并置换放射性标记配体的能力直接体现为放射性信号的降低,研究人员从而能够计算其半数抑制浓度(IC50)和抑制常数(Ki),以量化其结合亲和力。
|
| 细胞实验 |
蛋白质印迹程序[3]
所有细胞系和子宫或异种移植物肿瘤组织在RIPA裂解缓冲液和Halt蛋白酶抑制剂中裂解/均质化。降解试验中的ER蛋白水平通过标准蛋白质印迹、细胞内蛋白质或在WES或JESS仪器上进行的数字蛋白质分析进行测量。完整方法见补充扩展方法。 细胞生长抑制试验[3] 除非另有说明,否则在96孔板上以2000个细胞/孔的速度进行细胞生长抑制研究,使用3倍连续稀释的8点DRCs。在第5天,使用cell Titer Glo测量细胞存活率,并使用GraphPad分析CTG数据。活细胞成像增殖和剂量基质药物组合测定在补充扩展方法中进行了描述。 |
| 动物实验 |
未成熟大鼠子宫营养试验[3]
该模型按照先前描述的方法进行,使用出生后30天(PND 30)以下的未成熟雌性大鼠。PND 18的Sprague-Dawley (SD)大鼠分别接受30 mg/kg的维普地孕烷或10 mg/kg的AZD-9496(溶于PEG400/2% Tween80溶剂中),每日一次灌胃(p.o.),连续3天(qdx3);或单次皮下注射100 mg/kg的氟维司群(溶于10% w/v乙醇、10% w/v苯甲醇和15% w/v苯甲酸苄酯的混合溶剂中,并用蓖麻油定容至100% w/v,即EBB/蓖麻油)。每组使用5只动物。动物在末次给药后24小时或氟维司群/皮下注射组第4天处死并采集组织。测量子宫重量,液氮速冻后储存于-80°C。采用Western blot法测定雌激素受体(ER)水平。 MCF7原位异种移植模型[3] 将0.36 mg、90天缓释的17β-雌二醇缓释片皮下植入8至10周龄的NOD/SCID雌性小鼠体内。1至2天后,每只小鼠的一侧乳腺脂肪垫注射5 × 10⁶/200 µL MCF7细胞(ATCC)。细胞以25 × 10⁶个/mL的浓度,在50/50的RPMI-1640无酚红培养基/Corning Matrigel膜基质混合物中制备。当肿瘤平均体积达到 200 mm³ 时开始给药。口服联合用药时,先给予维普地孕烷,30 至 60 分钟后给予第二种药物。除非另有说明,所有口服药物(维普地孕烷、帕博西尼、阿贝西利、瑞博西尼、伊那维西布、阿培利西布和依维莫司)均以 5 mL/kg 的剂量每日一次给药,持续 28 天 (qdx28)。氟维司群皮下注射的给药方案为:4 mL/kg,每周两次 (biw),持续 2 周,然后每周一次 (qw),持续 2 周 (biwx2, qwx2)。体内给药的各种化合物的赋形剂列于补充表 S5 中。在疗效研究中,每周测量两次肿瘤体积,并使用公式 (宽度² × 长度)/2 计算,其中所有测量单位均为毫米 (mm),肿瘤体积单位为 mm³。每周记录两次体重。在一些药物联合疗效研究中,如果任何一组小鼠的体重下降接近 10%,则对所有组小鼠实施单日停药(图 6D 和 6E 中的黑色小箭头)。研究结束时,小鼠在末次给药后 18 小时处死,并将采集的组织用干冰速冻。肿瘤生长指数 (TGI) 按以下公式计算,肿瘤体积以 mm³ 表示: |
| 药代性质 (ADME/PK) |
用法与用量:Vepdegestrant 为口服给药,每日一次,随餐服用。推荐的 III 期单药治疗剂量为每日一次 200 mg。
吸收与血浆暴露:单次口服 200 mg 后,几何平均血药峰浓度(Cmax)为 630.9 ng/mL,24 小时药时曲线下面积(AUC0-24h)为 10,400 ng∙hr/mL。多次给药(每日一次)后,几何平均 Cmax 增至 1,056 ng/mL,24 小时 AUC 增至 18,310 ng∙hr/mL,表明重复给药存在中度蓄积。 代谢:Vepdegestrant 通过细胞色素 P450 3A4(CYP3A4)代谢。根据体外研究,未鉴定出人特异性代谢物,且大鼠的代谢谱与人类重叠。 药物相互作用:禁止与强 CYP3A4 抑制剂或诱导剂合用,或需调整剂量。临床试验中排除了正在使用强 CYP3A4 抑制剂或诱导剂的患者。如果允许既往使用,则 CYP3A4 抑制剂需在入组前停药 ≥7 天,强 CYP3A4 诱导剂需在入组前停药 ≥14 天。 蛋白结合与清除:在临床前研究中,肝细胞清除率与大鼠体内 PK 数据的相关性优于肝微粒体清除率。基于大鼠数据开发的生理药代动力学模型能够准确模拟 vepdegestrant 在进食和空腹状态下的 PK 特征。基于体外 ADME 数据构建的人体模型与口服给药后的血浆浓度曲线高度吻合。 跨物种与种族比较:在 200 mg 每日一次的剂量下,日本患者与西方患者之间未观察到显著的药代动力学差异。 Vepdegestrant是一种具有口服生物利用度的PROTAC药物。基于生理的药代动力学模型已从临床前物种(小鼠、大鼠、狗)的数据开发出来,以预测其在人体内的PK特征。这些模型整合了溶解度、渗透性和代谢(如肝脏微粒体固有清除率)等数据。临床前研究表明,肝细胞清除率比微粒体清除率更接近于体内PK数据。利用体外ADME数据构建的人体PBPK模型已成功预测了其PK特征,其口服吸收主要依赖于被动扩散。这些模型被用于支持临床试验中的剂量选择。 |
| 毒性/毒理 (Toxicokinetics/TK) |
剂量限制性毒性:在一项针对 ER+/HER2- 晚期乳腺癌日本患者接受 200 mg 每日一次给药的 I 期研究中,首个 28 天治疗周期内未观察到剂量限制性毒性。
不良事件:在该日本 I 期研究中,66.7% 的患者出现了治疗相关不良事件。除一例 2 级贫血外,所有治疗相关不良事件均为 1 级。未发生导致剂量降低或停药的不良事件。 III 期安全性特征:在一项比较 vepdegestrant(200 mg 每日一次)与氟维司群的 III 期试验中: 不良事件发生率:vepdegestrant 组为 86.9%,氟维司群组为 81.4% 。 vepdegestrant 最常见的不良事件为疲乏(26.6%)、天冬氨酸氨基转移酶升高(14.4%)和丙氨酸氨基转移酶升高(14.4%) 。 3-4 级不良事件发生率:vepdegestrant 组为 23.4%,氟维司群组为 17.6%。vepdegestrant 组最常见的 4 级事件为中性粒细胞减少症(1.9%)和低钾血症(1.9%) 。 9.9% 的 vepdegestrant 患者出现 QT 间期延长,但无临床后遗症 。 因不良事件导致的停药率较低,但 vepdegestrant 组(2.9%)高于氟维司群组(0.7%) 。 无死亡事件直接归因于任一治疗 。 临床前安全性数据:根据材料安全数据表,vepdegestrant 在标准标准下不属于危险物质或混合物。 CYP 相关毒性:由于 vepdegestrant 主要通过 CYP3A4 代谢,临床试验中排除了与强 CYP3A4 抑制剂或诱导剂的联合用药,以避免因暴露量改变而产生毒性。 根据安全数据表,vepdegestrant在科研用途中被归类为“非危险物质或混合物”。然而,全面的毒理学数据仍限于临床前模型和早期临床试验报告。I/II期临床研究表明其安全性良好,大多数不良事件可控。它禁用于已知有严重心血管疾病、药物性肺炎以及正在服用强效CYP3A抑制剂或诱导剂的患者。该产品严格限用于科研,不得在批准的临床试验之外用于人体治疗。 |
| 参考文献 | |
| 其他信息 |
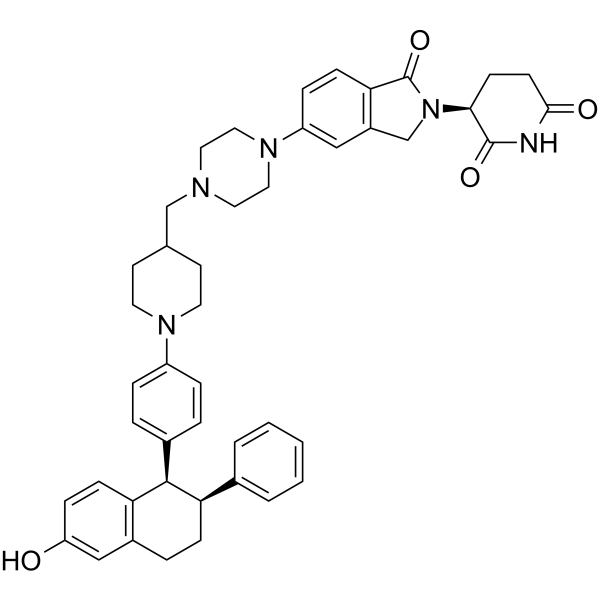
维普地孕烷是一种口服的异双功能分子,也是一种选择性雌激素受体 (ER) α 靶向蛋白降解剂,采用蛋白水解靶向嵌合体 (PROTAC) 技术制备,具有潜在的抗肿瘤活性。维普地孕烷由一个 ER α 配体和一个 E3 泛素连接酶识别基团组成。口服后,维普地孕烷靶向并结合 ER α 上的 ER 配体结合域。E3 泛素连接酶通过 E3 泛素连接酶识别基团募集到 ER 上,并使 ER α 被泛素化。这导致 ER α 被蛋白酶体泛素化和降解。这降低了 ER α 蛋白水平,减少了 ER α 靶基因的表达,并阻断了 ER 介导的信号传导。最终,抑制了 ER α 过表达肿瘤细胞的增殖。此外,ERα蛋白的降解会释放ARV-471,并可与其他ERα靶蛋白结合。ERα在多种癌症中过度表达,并在癌细胞增殖中发挥关键作用。
美国 FDA 已批准 VEPPANU 用于治疗既往接受过至少一线内分泌治疗后出现疾病进展的 ER+/HER2-、ESR1 突变的晚期或转移性乳腺癌成人患者。这是 FDA 首次批准 PROTAC 蛋白降解剂疗法。在关键的 III 期 VERITAC-2 试验中,对于携带 ESR1 突变的患者,与氟维司群相比,vepdegestrant 将疾病进展或死亡风险显著降低了 43%(中位 PFS:5.0 个月 vs. 2.1 个月;HR 0.57)。大多数不良事件为低级别(1-2 级)。该批准填补了这类侵袭性乳腺癌患者未被满足的重大医疗需求。 |
| 分子式 |
C45H49N5O4
|
|---|---|
| 分子量 |
723.9017
|
| 精确质量 |
723.37845
|
| 元素分析 |
C, 74.66; H, 6.82; N, 9.67; O, 8.84
|
| CAS号 |
2229711-68-4
|
| 相关CAS号 |
2229711-08-2 (racemate)
|
| PubChem CID |
134562533
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
6.4
|
| tPSA |
96.4Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
54
|
| 分子复杂度/Complexity |
1310
|
| 定义原子立体中心数目 |
3
|
| SMILES |
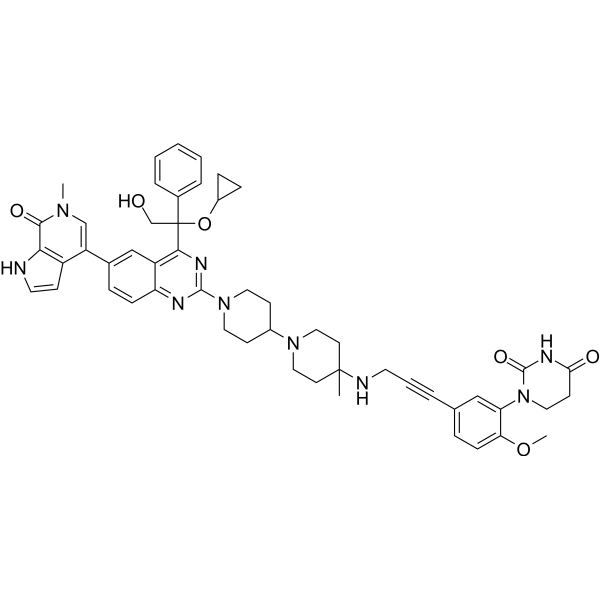
O([H])C1C([H])=C([H])C2=C(C=1[H])C([H])([H])C([H])([H])[C@]([H])(C1C([H])=C([H])C([H])=C([H])C=1[H])[C@]2([H])C1C([H])=C([H])C(=C([H])C=1[H])N1C([H])([H])C([H])([H])C([H])(C([H])([H])N2C([H])([H])C([H])([H])N(C3C([H])=C([H])C4C(N([C@]5([H])C(N([H])C(C([H])([H])C5([H])[H])=O)=O)C([H])([H])C=4C=3[H])=O)C([H])([H])C2([H])[H])C([H])([H])C1([H])[H]
|
| InChi Key |
TZZDVPMABRWKIZ-XMOGEVODSA-N
|
| InChi Code |
InChI=1S/C45H49N5O4/c51-37-12-15-39-33(27-37)8-13-38(31-4-2-1-3-5-31)43(39)32-6-9-35(10-7-32)48-20-18-30(19-21-48)28-47-22-24-49(25-23-47)36-11-14-40-34(26-36)29-50(45(40)54)41-16-17-42(52)46-44(41)53/h1-7,9-12,14-15,26-27,30,38,41,43,51H,8,13,16-25,28-29H2,(H,46,52,53)/t38-,41+,43+/m1/s1
|
| 化学名 |
(3S)-3-[6-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl]phenyl]piperidin-4-yl]methyl]piperazin-1-yl]-3-oxo-1H-isoindol-2-yl]piperidine-2,6-dione
|
| 别名 |
ARV-471; 2229711-68-4; Vepdegestrant; ARV-471 (protac); WC1U3R1YMI; ARV471; (S)-3-(5-(4-((1-(4-((1R,2S)-6-Hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl)phenyl)piperidin-4-yl)methyl)piperazin-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮和光照。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 110 mg/mL (151.95 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 5.5 mg/mL (7.60 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 55.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 配方 2 中的溶解度: ≥ 2 mg/mL (2.76 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.0 mg/mL 澄清的 DMSO 储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 View More
配方 3 中的溶解度: 2 mg/mL (2.76 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.3814 mL | 6.9070 mL | 13.8141 mL | |
| 5 mM | 0.2763 mL | 1.3814 mL | 2.7628 mL | |
| 10 mM | 0.1381 mL | 0.6907 mL | 1.3814 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT01042379
Conditions:Breast Neoplasms|Breast Cancer|Breast Tumors|Angiosarcoma|TNBC - Triple-Negative Breast Cancer|HER2-positive Breast Cancer|HER2-negative Breast Cancer|Hormone Receptor Positive Tumor|Hormone Receptor Negative Tumor|Early-stage Breast Cancer|Locally Advanced Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT06125522
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT04606446
Conditions:Locally Advanced or Metastatic ER+ HER2- Breast Cancer|Locally Advanced or Metastatic Castration-resistant Prostate Cancer|Locally Advanced or Metastatic Non-small Cell Lung Cancer
Title:A Phase 1/2 Trial of ARV-471 Alone and in Combination With Palbociclib (IBRANCE®) in Patients With ER+/HER2- Locally Advanced or Metastatic Breast Cancer
Status:Completed
updateDate:2026-04-15
Ctid:NCT04072952
Link: https://clinicaltrials.gov/ct2/show/NCT04072952
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT07231991
Conditions:Healthy ParticipantsLink: https://clinicaltrials.gov/ct2/show/NCT05654623
Conditions:Advanced Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT05573555
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT06206837
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT05909397
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT05548127
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT05549505
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT06911788
Conditions:Healthy ParticipantsLink: https://clinicaltrials.gov/ct2/show/NCT05732428
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT06256510
Conditions:Healthy ParticipantsLink: https://clinicaltrials.gov/ct2/show/NCT05463952
Conditions:Breast NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT05538312
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT05673889
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT05652660
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT06275841
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT06005688
Conditions:Healthy ParticipantsInvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
