| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| Other Sizes |
|

| 靶点 |
N-methyl-D-aspartate (NMDA) receptor subunit 2B (GluN2B) [Ki = 8.1 nM; IC50 = 3.6 nM)
|
|---|---|
| 体外研究 (In Vitro) |
对于激动剂刺激的 NMDA-GluN1a/GluN2BL(tk-) 细胞,rislenemdaz (CERC-301) 可减少钙流入,IC50 为 3.6 nM。当 GluN2B 受体与所有其他靶标(包括 hERG 钾通道)进行比较时,rislenemdaz 表现出至少 1000 倍的选择性。 10 uM 的 rislenemdaz 对 sigma 型受体的作用最小[1]。
体外药理[1] Rislenemdaz/CERC‐301对NMDA‐GluN2B受体的亲和力 [1] CERC‐301有效抑制放射性配体([3H]Compound‐2)与人类NMDA‐GluN1a/GluN2B受体的结合,这些受体在所有被测试物种(大鼠、狗、恒河猴、人)的L(tk‐)细胞和脑组织浆液中表达。利用人颞叶皮层匀浆测定CERC‐301的结合亲和力,在室温和37℃下的K i值分别为3.1 nmol L−1 (0.0031 μmol L−1)和8.1 nmol L−1 (0.0081 μmol L−1)。在其他物种中的发现与人类数据一致(表1)。 Rislenemdaz/CERC‐301对NMDA受体的功能活性和选择性 [1] CERC‐301抑制钙流入激动剂刺激的NMDA‐GluN1a/GluN2B L(tk‐)细胞,IC50为3.6 nmol L−1,但当浓度高达30,000 nmol L−1 (30 μmol L−1)时,对钙流入激动剂刺激的GluN1a/GluN2A细胞没有影响。 Rislenemdaz/CERC‐301对NMDA‐GluN2B受体影响的电生理学研究 [1] CERC - 301的效价通过GluN2B拮抗剂电压箝位法测定。在10 μmol L−1谷氨酸和10 μmol L−1甘氨酸的存在下,测量了CERC‐301的开启率(图2A)和关闭率(图2B)。这些实验表明,k开和k关率分别为1.3 × 105 mol L−1 s−1和~2 × 10−5 s−1,kd约为0.15 nmol L−1。 Rislenemdaz/CERC‐301的反筛选 [1] 当浓度等于或大于10 μmol L−1 (~3584 ng mL−1)时,CERC‐301在酶和放射性配体结合试验中没有表现出显著的活性。CERC‐301对GluN2B受体对所有测试靶标(包括hERG钾通道)表现出至少1000倍的选择性。CERC‐301在10 μmol L−1 (sigma‐1、sigma‐2和非特异性)时对sigma‐型受体也表现出最小的活性。 |
| 体内研究 (In Vivo) |
与赋形剂对照相比,Rislenemdaz (CERC-301)(1、3、10 和 30 mg/kg)显着改善游泳行为(10 mg/kg 时 P<0.05;1、3 和 30 mg/kg 时 P<0.01) )并大大降低了不动的频率(P<0.001)。采样时大鼠血浆中rislenemdaz的水平约为15、120、390、1420、4700和14,110 nM (0.015、0.120、0.390、1.42、4.7和14.11 uM)。这分别相当于约 5%、29%、56%、83%、94% 和 98% RO。大约 0.3 和 0.7 mg/kg,或大约 RO 的 30% 和 50%,分别是增加游泳频率和减少不动的估计 ED50。与载体对照(1 mg/kg P < 0.01;3、10 和 30 mg/kg P < 0.001)的总行驶距离相比,rislenemdaz(1、3、10 和 30 mg/kg)显着增加了距离旅行。很少甚至没有测试[1]。
急性抑郁模型[1] 强迫游泳试验[1] Rislenemdaz(1,3,10和30 mg kg - 1)显著降低不动频率(P < 0.001),显著增加游泳行为(P < 0.01);与车辆对照组相比,10 mg kg - 1的P < 0.05)(图3),但除了3 mg kg - 1的剂量外,不影响攀爬行为(P < 0.05)。与车辆对照组相比,地西帕明(20 mg kg−1)显著降低了静止不动(P < 0.001),显著提高了攀爬行为(P < 0.01),游泳行为没有变化。采样时,CERC‐301的血浆水平分别约为15、120、390、1420、4700和14110 μmol L−1(0.015、0.120、0.390、1.42、4.7和14.11 μmol L−1),对应于大鼠的RO分别约为5、29、56、83、94和98%。游泳频率增加和不活动减少的ED50分别为~0.3和0.7 mg kg - 1,对应的RO为~ 30%和50%。 运动试验[1] Rislenemdaz(1,3,10和30 mg kg - 1)显著增加了旅行距离(P < 0.01);在测试的前5分钟(时间与强迫游泳测试的时间相关),与车辆对照组相比,10和30 mg kg - 1的P < 0.001。CERC‐301(1、3、10和30 mg kg - 1)显著增加总旅行距离(P < 0.01);3、10和30 mg kg−1)与60分钟试验期间的对照相比,P < 0.001。运动活动增加的ED50为~2 mg kg−1,转化为~75%的RO,高于游泳频率增加和不动减少的ED50。0.1和0.3 mg kg - 1剂量组未观察到运动影响(图3)。 遥测大鼠血流动力学研究[1] 单次口服Rislenemdaz/CERC - 301 /CERC - 301,在0.3 ~ 1 mg kg - 1 (ED50≈0.7 mg kg - 1)剂量范围内,清醒大鼠动脉血压呈剂量依赖性短暂升高,且该效应在1 ~ 10 mg kg - 1剂量范围内达到稳定(图4)。在给药后1 - 2小时观察到峰值效应(图4A),与大鼠CERC‐301的PK谱一致。在0.2 mg kg - 1(例如,收缩压+36 mmHg)时,CERC‐301的血流动力学变化幅度小于MK‐801。CERC‐301给药后3.5小时内的血流动力学变化与活动水平呈线性相关(根据遥测记录估计);特别是,活性是HR的预测因子(r2 = 0.67),因此,CERC‐301的兴奋作用可能解释了在大鼠中观察到的HR增加(特别是在高剂量水平下)。该研究还表明α1‐和/或β1‐AR阻断剂可能分别对CERC‐301介导的血压升高(图4B)和心率升高(数据未显示)提供保护。 |
| 酶活实验 |
体外药理测定 [1]
Rislenemdaz/CERC‐301对NMDA‐GluN2B受体的亲和力[1] 如前所述,在室温(或37°C)下进行放射性配体结合试验(Kiss等人,2005;Liverton et al. 2007)。类似的结合实验,如附录S1所述,克隆的人类受体在L(tk‐)细胞中表达,也使用全脑匀浆(大鼠)、额叶皮质匀浆(狗和猕猴)和颞叶皮质匀浆(人)进行。更详细的描述见附录S1。 Rislenemdaz/CERC‐301对NMDA受体的功能活性和选择性 [1] 我们测量了表达GluN1a/GluN2B或GluN1a/GluN2A人受体的L(tk‐)细胞对钙内流的抑制,以确定CERC‐301抑制NMDA受体功能的IC50,如前所述(Kiss et al. 2005;另见附录S1)。 Rislenemdaz/CERC‐301的电生理研究 [1] 利用克隆的人类NMDA‐GluN2B受体进行体外电生理测定,以确定CERC‐301的结合和解离动力学以及效力(Kiss et al. 2005;另见附录S1)。 Rislenemdaz/CERC‐301的反筛谱 [1] CERC‐301在一系列标准受体结合和酶分析中进行评估。CERC - 301在体外以10、30或100 μmol L - 1(分别为~3584、10752或35840 ng mL - 1)检测其与参考配体竞争的能力,以评估可能未检测到的药理活性(研究了bbb150受体和酶,包括sigma型阿片受体)。 非临床药代动力学[1] 血浆蛋白结合[1] 大鼠、狗、恒河猴和人血浆样品(3 mL, N = 3)分别加入2和20 μmol L−1[14℃]CERC‐301,在37℃的摇水浴中孵育30 min。孵育后,采用标准超离心方法测定药物未结合的百分比(Pacifici和Viani 1992)。 |
| 动物实验 |
大鼠神经毒性[1]
\n将24只Sprague-Dawley大鼠(雌雄各12只)分为四组,分别单次灌胃给予赋形剂(0.5%甲基纤维素[MC]和0.02%十二烷基硫酸钠[SLS]的去离子水)或Rislenemdaz/CERC-301,剂量分别为10、30或100 mg kg−1,灌胃体积为10 mL kg−1。另取12只雄性大鼠,单次皮下注射MK-801(NMDA受体的非竞争性拮抗剂;阳性对照),剂量为10 mg kg−1,注射体积为2 mL kg−1。每组雌雄各6只大鼠在给药后4至6小时处死并进行尸检,每组剩余大鼠在给药后3天(第4天)处死并进行尸检。活体观察和测量包括体重和临床观察。处死时,大鼠被麻醉并进行灌注固定。尸检时,收集脑组织进行组织病理学评估。\n \nRislenemdaz/CERC-301和MK-801评估组的动物在预定的尸检时间间隔(给药后4-6小时或第4天)处死。所有动物均使用异氟烷/氧气混合物麻醉,并通过左心室灌注肝素化的0.001%亚硝酸钠生理盐水。生理盐水冲洗后,灌注10%中性缓冲福尔马林(NBF)。取出脑组织,称重后保存在10%中性缓冲福尔马林溶液(NBF)中。\n \n将脑组织切成2毫米厚的冠状切片,每只动物取三个组织块,每个组织块包含多个切片。对以下脑区进行染色:新皮层、古皮层、基底核、边缘系统、丘脑/下丘脑、中脑、小脑、脑桥和延髓。所有在给药后4至6小时处死的动物以及所有在给药后3天处死的动物的脑组织切片均进行石蜡包埋,切片厚度为5微米,并用苏木精-伊红染色,最后进行显微镜检查。对于在第 4 天(给药后 3 天)处死的大鼠,将 1 号和 2 号块的连续切片用 Fluoro-Jade B(一种可提高评估大脑神经元变性敏感性的染色剂)和胶质纤维酸性蛋白(一种用于星形胶质细胞反应的染色剂)染色,并在显微镜下进行检查。另外三组大鼠(每组四只雄性和三只雌性)以相同方式口服CERC-301,并连续24小时采集血样,分析血浆中利塞那唑/CERC-301的浓度,并评估其全身暴露量。\n \n\n体内药理学[1] \nGluN2B受体占有率与血浆药物浓度的相关性[1] \n如附录S1和先前文献(Liverton等人,2007)所述,CERC-301/利塞那唑已分别给予大鼠、犬和恒河猴。此外,还测定了大鼠静脉注射[3H]化合物-3后的受体占有率。如附录 S1 所述,在静脉注射 (IV) 后 15 分钟和口服 (PO) 后 60 分钟评估了血浆浓度与脑组织 RO 之间的关系。\n \n\n急性抑郁模型 [1] \n强迫游泳试验 [1] \n将年轻、成年雄性 Sprague-Dawley 大鼠随机分配到各治疗组,并分别给予赋形剂(0.5% MC/0.02% SLS)、溶于无菌水的参考化合物地昔帕明(20 mg kg−1;一种三环类抗抑郁药)或溶于 0.5% MC/0.02% SLS 的利昔拉明/CERC-301(0.1、0.3、1、3、10 和 30 mg kg−1),在第 1 天给药两次(适应后;试验前约 24 小时,黑暗周期前),并在第 2 天给药一次。 2(地昔帕明组进行30分钟预测试,CERC-301和溶剂对照组进行45分钟预测试)。\n \n每个强迫游泳室均由透明亚克力制成(高40厘米,直径20.3厘米)。大鼠在装有23°C ± 1°C水的圆柱体中进行一次15分钟的给药前游泳测试,约24小时后进行5分钟的实验测试。适应性测试期间水深为16厘米,测试期间水深为30厘米。每5秒记录一次不动、攀爬和游泳行为,每只动物共记录60次。当动物无法保持鼻子露出水面的姿势时,立即将其从水中取出并排除出实验。在游泳试验结束后采集血液样本,并分析血浆中利塞那唑/CERC-301的浓度。\n \n\n运动活性测定[1] \n为了确认利塞那唑/CERC-301在强迫游泳试验中的作用并非由于活动水平的普遍增加所致,在口服CERC-301后对大鼠进行了运动活性测定。将42只成年雄性Sprague-Dawley大鼠随机分配到各处理组(载体组或CERC-301组,剂量分别为0.1、0.3、1、3、10和30 mg kg−1;每组n = 6)。在光照周期内,于光电管监测笼中评估大鼠的运动活性。每个笼子由一个标准塑料鼠笼(24 × 45.5 cm)和一个不锈钢框架组成。红外光电管光束沿框架长轴方向布置,用于测量动物的活动距离。另一组光束置于地面上方,用于测量动物的站立活动。计算机系统记录光电管光束的中断情况。测试期间,在测试笼顶部放置滤光片。大鼠在第1天(测试前约24小时,黑暗周期开始前)通过灌胃法分别给予两次赋形剂或测试化合物,并在第2天(放入运动笼进行60分钟测试前45分钟)给予一次。运动活动以5分钟为单位进行记录。\n \n\n遥测大鼠的血流动力学效应[1] \n为了确定全身血流动力学效应,对6只(n = 6)慢性遥测大鼠(植入时间至少在给药前7天)单次灌胃给予Rislenemdaz/CERC-301,剂量分别为0.3、0.6、1、3和10 mg kg−1,并记录全身血压和心率值。将这些血流动力学效应与静脉注射0.2 mg kg−1剂量的MK-801(溶于0.9%生理盐水)进行比较。每只动物均单次灌胃给予Rislenemdaz/CERC-301或赋形剂(0.5% MC/0.02% SLS)(体积:5 mL kg−1)。在给药前(仅给予赋形剂)和每次口服给药后进行24小时记录。在另一组研究中,将CERC-301(1 mg kg−1)与阿替洛尔(β1-肾上腺素能阻滞剂,1 mg kg−1,静脉推注)和哌唑嗪(α1-肾上腺素能受体拮抗剂,200 μg kg−1,静脉推注)联合给药,以阐明高血压的潜在机制。数据分析并与基线进行比较,并校正了给药前24小时的载体对照。\n \n\n首次人体药代动力学研究[1] \n该研究方案已获得机构审查委员会的批准,所有受试者均签署了书面知情同意书。24名健康的年轻男性受试者被随机分配到3个连续治疗组(A、B和C)之一。每个治疗组包含8名受试者,其中2名受试者接受安慰剂,6名受试者接受单次递增口服剂量的Rislenemdaz/CERC-301,每次给药之间至少间隔7天的洗脱期:A组(0.1、0.2、0.5、1和2 mg);B组(2、4、8和15 mg,以及4 mg与食物同服);C组(15和20 mg)。分别于给药前及给药后0.5、1、1.5、2、3、4、6、9、12、18、24、30、48和72小时采集血样。采用反相高效液相色谱-串联质谱法分析血浆样本中CERC-301的浓度。分析范围为0.5至500 nmol L⁻¹(0.180至180 ng mL⁻¹)。 |
| 药代性质 (ADME/PK) |
人体药代动力学研究[1]
CERC‐301/Rislenemdaz吸收迅速(图5A),在所有空腹剂量水平下,平均达峰时间(Tmax)均在给药后1小时内达到,4至20 mg剂量范围内的末端消除半衰期约为12至17小时。在所研究的剂量范围内,Cmax和AUC呈剂量比例关系。餐后给药导致Tmax延迟约1小时,Cmax降低约56%,但总体吸收程度(AUC)不受影响(图5B,表3)。 血浆蛋白结合[1] CERC–301/Rislenemdaz在37°C下对大鼠(89.6%)、狗(97.2%)、猴子(96.9%)和人类(97.7%)表现出高度的浓度非依赖性血浆蛋白结合。 |
| 毒性/毒理 (Toxicokinetics/TK) |
非临床安全性[1]
大鼠神经毒性[1] 在大鼠中,单次给予Rislenemdaz/CERC‐301(10、30 和 100 mg kg−1)和溶剂对照,均未在所有检测的脑区引起空泡化或坏死。在这些剂量下,平均 Cmax 分别约为 4、14 和 26 μmol L−1(1433、5018 和 9319 ng mL−1)。相比之下,所有给予 MK‐801(10 mg kg−1)的动物的扣带回神经元均出现空泡化和坏死,这与之前的报道一致(Fix 等,1995)。在4-6小时时间点,所有接受MK-801治疗的动物(6只雄性;第5组)的大脑皮层扣带回区域第3层和第4层均出现大量空泡化神经元。受影响的神经元的特征是胞质内充满大量紧密排列、界限分明的空泡。在第4天,所有接受MK-801治疗的动物(6只雄性;第5组)的大脑皮层扣带回区域第3层和第4层均出现坏死神经元(使用Fluoro-Jade B染色剂显色),这与之前的报道一致(Fix等,1995)。在MK-801治疗的动物中,胶质纤维酸性蛋白(GFAP)免疫组化染色显示扣带回区域的染色强度略有增加。 |
| 参考文献 | |
| 其他信息 |
Mk 0657 已用于研究重度抑郁症治疗的试验。
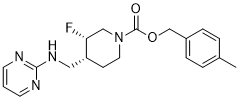
对口服生物利用度高的选择性 N-甲基-D-天冬氨酸 (NMDA) 受体亚基 2B (GluN2B) 拮抗剂 4-甲基苄基 (3S, 4R)-3-氟-4-[(嘧啶-2-基氨基)甲基]哌啶-1-羧酸酯 (CERC-301/利赛尼达) 进行了临床前药效学和药代动力学表征,旨在开发一种基于受体占有率 (RO) 的转化方法,以指导 CERC-301 在重度抑郁症临床试验中的剂量选择。CERC-301 对 GluN2B 具有高亲和力 (Ki,8.1 nmol L-1),IC50 为 3.6 nmol L-1,且无脱靶活性。 CERC-301 在强迫游泳试验中显示出疗效,有效剂量 (ED50) 为 0.3-0.7 mg kg⁻¹(RO,30-50%);在 ED50 为 2 mg kg⁻¹ 时观察到运动活性增加,对应的 RO 为 75%。预测的人体 50% RO 浓度 (Occ50) 为 400 nmol L⁻¹,与大鼠、犬和猴的预测值(分别为 300、200 和 400 nmol L⁻¹)相似。安全性药理学和神经毒性研究未发现任何特定的安全性问题。一项在健康男性中进行的首次人体研究表明,其药代动力学特征与剂量呈正比,Tmax 约为 1 小时,t1/2 为 12-17 小时。基于临床前和药效学数据,推测在人体中,≥8 mg 的剂量具有可接受的安全性,并能达到临床相关的血浆峰值暴露量。[1] 在清醒遥测大鼠中的研究表明,口服利赛仑达兹/CERC-301 可短暂且呈剂量依赖性地升高动脉血压,剂量范围为 0.3 至 1 mg kg⁻¹,并在 1 至 10 mg kg⁻¹ 时达到平台期。有趣的是,血压效应的 ED50 与强迫游泳试验的 ED50 相似。CERC-301 引起的血流动力学变化幅度小于广谱 NMDA 受体拮抗剂 MK-801。MK-801 的研究结果与先前发表的关于 MK-801 的机制数据(Lewis 等,1989)一致。心率和血压的变化可能部分归因于药物依赖性增强大鼠的运动能力;在运动功能研究中也观察到了剂量依赖性运动分析,并在较高剂量下呈现出支持这一趋势。本研究还表明,α1-肾上腺素能阻断剂可能对CERC-301介导的血压升高具有保护作用;然而,还需要更多研究来进一步阐明其潜在机制。[1] 与一些临床使用的NMDA受体拮抗剂(例如氯胺酮或美金刚)不同,CERC-301在单次口服剂量高达100 mg kg−1的大鼠中未显示出神经毒性。这一结果与其他GluN2B特异性受体拮抗剂(例如CP-101,606和Ro 63-1908)的研究结果一致,这些拮抗剂均未表现出神经毒性。其中,CP-101,606甚至可能对发育中的大脑具有神经保护作用(Gill等人,2002;Lewis等人,2012)。GluN2B特异性拮抗剂在非临床环境中未表现出神经毒性信号,且在临床环境中未观察到类似氯胺酮的精神活性作用,因此,如果未来的临床试验数据需要,可以增加这些药物的剂量以达到更高的受体占有率(高于本文预测的水平)。 [1] CERC-301 首次人体研究的药代动力学数据显示,在 0.1 至 20 mg 单次剂量(空腹)范围内,平均 Cmax 为 0.007 μmol L−1 (2.4 ng mL−1) 至 1.65 μmol L−1 (590.3 ng mL−1),其半衰期使其适合每日一次给药。[1] CERC-301 是一种强效、口服生物利用度高的选择性 NMDA-GluN2B 受体拮抗剂,具有抗抑郁作用。基于临床前药代动力学和药效学数据,推测每日 8 mg 的慢性给药方案具有可接受的安全性,可达到临床相关的血浆和脑组织浓度,并能快速发挥抗抑郁作用。[1] |
| 分子式 |
C19H23FN4O2
|
|---|---|
| 分子量 |
358.409927606583
|
| 精确质量 |
358.18
|
| 元素分析 |
C, 63.67; H, 6.47; F, 5.30; N, 15.63; O, 8.93
|
| CAS号 |
808732-98-1
|
| 相关CAS号 |
808733-06-4 (HCl);808732-98-1;1893392-76-1 (mesylate);
|
| PubChem CID |
11394238
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
527.4±60.0 °C at 760 mmHg
|
| 闪点 |
272.7±32.9 °C
|
| 蒸汽压 |
0.0±1.4 mmHg at 25°C
|
| 折射率 |
1.584
|
| LogP |
2.73
|
| tPSA |
67.4
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
439
|
| 定义原子立体中心数目 |
2
|
| SMILES |
CC1=CC=C(C=C1)COC(=O)N2CC[C@@H]([C@@H](C2)F)CNC3=NC=CC=N3
|
| InChi Key |
RECBFDWSXWAXHY-IAGOWNOFSA-N
|
| InChi Code |
InChI=1S/C19H23FN4O2/c1-14-3-5-15(6-4-14)13-26-19(25)24-10-7-16(17(20)12-24)11-23-18-21-8-2-9-22-18/h2-6,8-9,16-17H,7,10-13H2,1H3,(H,21,22,23)/t16-,17-/m1/s1
|
| 化学名 |
4-methylbenzyl (3S,4R)-3-fluoro-4-((pyrimidin-2-ylamino)methyl)piperidine-1-carboxylate
|
| 别名 |
CERC-301; CERC301; Rislenemdaz; 808732-98-1; Rislenemdaz [USAN]; Rislenemdaz, (-)-; MK-0657, (-)-; 4-Methylbenzyl (3S,4R)-3-fluoro-4-((pyrimidin-2-ylamino)methyl)piperidine-1-carboxylate; CERC 301; MK-0657; MK 0657; MK0657;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~279.01 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.98 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.98 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.98 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7901 mL | 13.9505 mL | 27.9010 mL | |
| 5 mM | 0.5580 mL | 2.7901 mL | 5.5802 mL | |
| 10 mM | 0.2790 mL | 1.3951 mL | 2.7901 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
(A) Time course ofN‐methyl‐D‐aspartate (NMDA) receptor inhibition by CERC‐301 at three concentrations of 30, 100, and 300nmolL−1.Pharmacol Res Perspect. 2015 Dec; 3(6): e00198. |
|---|
In‐vivo efficacy and potential central nervous system (CNS) side effects of CERC‐301 when orally administered in rats. Efficacy is depicted by a reduction in immobility frequency (filled circles; left axis) during the forced swim test. Potential CNS side effect is depicted by an increase in total distance traveled (open squares; right axis) as a function of dose.Pharmacol Res Perspect. 2015 Dec; 3(6): e00198. |
Effects of a single oral dose ofCERC‐301 on systolic blood pressure.Pharmacol Res Perspect. 2015 Dec; 3(6): e00198. |
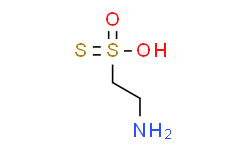 Thiotaurine
Thiotaurine
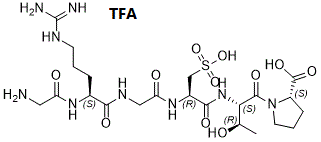 ALG1001 TFA
ALG1001 TFA
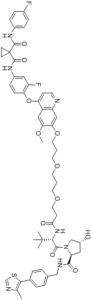 SJF-8240 (Foretinib-Based PROTAC 7)
SJF-8240 (Foretinib-Based PROTAC 7)
 BMS-066
BMS-066
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
