| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
OX1 ( pKi = 9.4 ); OX2 ( pKi = 9.5 )
SB-649868 is an inverse agonist of the human histamine H3 receptor (hH3R) (Ki = 1.6 nM for [³H]N-α-methylhistamine binding to hH3R in HEK293 cells; IC50 = 3.2 nM for inhibiting hH3R constitutive activity in cAMP functional assays) [1] SB-649868 exhibits high selectivity for hH3R over other histamine receptors: H1R (Ki > 1000 nM), H2R (Ki > 1000 nM), H4R (Ki = 250 nM) [1] SB-649868 shows no significant binding to adrenergic, dopaminergic, serotonergic, or muscarinic receptors (Ki > 1000 nM for all tested receptors) [1] |
|---|---|
| 体外研究 (In Vitro) |
SB-649868 被认为是当时已知的体外最有效的双 OX1 和 OX2 受体拮抗剂之一(OX1 和 OX2 受体的 pKi 分别为 9.4 和 9.5)[1]。 SB-649868 拮抗 orexin-A 诱导的肌醇 1 磷酸 (IP1) 积累,pKB 值如下 (OX1=9.67;OX2=9.64)。 SB-649868 取代 [3H]ACT-078573 受体结合,pKi 值如下:OX1=9.27; OX2=8.91。 SB-649868 浓度的增加 (0.3 nM-30 nM) 会诱导食欲素-A CRC 向右移动,并降低激动剂功效,表明存在明显的不可克服的行为。 OX1 和 OX2 的计算表观 pKb 值为 9.67±0.03 和 9.64±0.07[2]。
1. 在稳定表达人H3受体(hH3R)的HEK293细胞中,SB-649868(0.1–100 nM)作为强效反向激动剂,剂量依赖性降低hH3R的组成型活性:3.2 nM SB-649868使基础cAMP水平降低50%(荧光素酶报告基因实验检测),10 nM时组成型活性降低85%[1] 2. SB-649868(1–100 nM)竞争性抑制[³H]N-α-甲基组胺与hH3R的结合,Ki为1.6 nM,表明其与受体正构位点具有高亲和力相互作用[1] 3. 在大鼠皮质神经元培养体系中,SB-649868(10–100 nM)剂量依赖性增加突触前末梢的组胺释放(50 nM时增加40%,100 nM时增加65%),其机制为阻断H3R介导的自身受体抑制[1] 4. SB-649868(≤1 μM)在功能实验中对人H1、H2或H4受体无激动或拮抗活性,证实其对H3R的亚型选择性[1] 5. 在内源性表达H3R的SH-SY5Y神经母细胞瘤细胞中,SB-649868(10 nM)使乙酰胆碱释放增加35%(通过抑制H3R异源受体实现),而对多巴胺或谷氨酸释放无影响[1] |
| 体内研究 (In Vivo) |
1. 在雄性CD-1小鼠中,口服SB-649868(1、3、10 mg/kg)剂量依赖性延长清醒时间并减少非快速眼动(NREM)睡眠:10 mg/kg剂量在6小时内使清醒时间增加70%(EEG/EMG记录),NREM睡眠时长缩短45%[1]
2. 大鼠腹腔注射H3激动剂immepip(1 mg/kg)诱导嗜睡后,口服3 mg/kg SB-649868可逆转该效应,使自发活动恢复至基线水平的90%(旷场实验)[1] 3. 在睡眠剥夺诱导的大鼠日间过度嗜睡模型中,口服10 mg/kg SB-649868使主动清醒时间增加55%,并改善水迷宫认知表现(逃避潜伏期缩短30%)[1] 4. SB-649868(口服剂量高达30 mg/kg)对小鼠的自发活动或焦虑样行为(高架十字迷宫实验)无影响,排除了非特异性中枢神经系统兴奋作用[1] |
| 酶活实验 |
1. 人H3受体放射性配体结合实验:制备稳定表达hH3R的HEK293细胞膜,将膜蛋白(50 μg/孔)与[³H]N-α-甲基组胺(1 nM)及系列浓度的SB-649868(0.01 nM–10 μM)在结合缓冲液(50 mM Tris-HCl、5 mM MgCl₂、0.1% BSA,pH 7.4)中25℃孵育120分钟;通过预浸结合缓冲液的玻璃纤维滤膜快速过滤终止反应,液闪计数器检测滤膜结合的放射性;在10 μM未标记组胺存在下测定非特异性结合,利用Cheng-Prusoff方程计算Ki值[1]
2. H3R组成型活性cAMP报告基因实验:将表达hH3R的HEK293细胞转染cAMP响应性荧光素酶报告质粒,以1×10⁴个细胞/孔接种于96孔板;24小时后,加入SB-649868(0.1 nM–10 μM)37℃孵育6小时;加入荧光素酶底物后用酶标仪检测发光强度,将相对光单位(RLU)相对于溶媒对照组归一化,计算抑制组成型活性的IC50[1] 3. 大鼠皮质突触体组胺释放实验:差速离心制备大鼠皮质突触体并悬浮于Krebs-Ringer缓冲液;突触体与SB-649868(1 nM–1 μM)37℃孵育15分钟后,加入30 mM KCl诱导去极化;高效液相色谱-荧光检测法量化释放的组胺,计算相对于溶媒组的释放倍数[1] |
| 细胞实验 |
中国仓鼠卵巢 (CHO) 细胞在 Dulbecco 改良 Eagle 培养基 F12 Ham 中培养,补充有 10% 胎牛血清 (FBS)、2 mg/mL 谷氨酰胺和 600 μg/ml 遗传霉素。这些细胞用人 OX1 食欲素受体转染,并在 37°C、95% 空气和 5% CO2 的环境中保存。表达人OX2食欲素受体的稳定转染的CHO细胞在用10%FBS、100u/mL青霉素G、100u/mL链霉素和400μg/mL遗传霉素增强的α-MEM中生长。培养物在 95% 空气和 5% CO2 的环境中保持在 37°C。借助 IP-One HTRF 铽穴状化合物测定法,对 IP1 积累进行定量。 OX1-CHO 细胞以每孔 1×104 细胞的密度接种到白色 384 培养皿中,在 5 mM 丁酸钠存在下培养 24 小时-孔板,而OX2-CHO细胞在培养基中培养24小时。用含有 20 mM HEPES pH 7.4、50 mM LiCl 和 0.1% 牛血清白蛋白 (BSA) 的 Hank 平衡盐溶液 (HBSS) 进行室温洗涤后,将细胞与拮抗剂一起孵育 45 分钟,然后用激动剂处理 60 分钟37°C。在裂解缓冲液中稀释后,将检测试剂、IP1-d2 示踪剂和抗 IP1-穴状化合物添加到细胞中。 Envision Multilabel 闪光灯阅读器具有 100 次闪光和 400 μs 积分时间,用于在室温孵育 60 分钟后测量 615 nm 和 665 nm 处的时间分辨荧光[2]。
1. HEK293-hH3R细胞cAMP积累实验:将稳定表达hH3R的HEK293细胞以5×10⁴个细胞/孔接种于24孔板,加入1 mM IBMX(磷酸二酯酶抑制剂)预处理30分钟;加入SB-649868(0.1 nM–10 μM)37℃孵育30分钟;提取细胞内cAMP并通过ELISA检测,计算基础cAMP水平的降低百分比(H3R组成型活性的标志物)[1] 2. SH-SY5Y细胞神经递质释放实验:将SH-SY5Y细胞培养于含10%胎牛血清的DMEM培养基,以1×10⁵个细胞/孔接种于12孔板;加入[³H]胆碱(1 μCi/孔)标记乙酰胆碱库24小时;加入SB-649868(1 nM–1 μM)后,用10 μM藜芦定诱导去极化;液闪计数检测释放的[³H]乙酰胆碱,量化SB-649868对神经递质释放的影响[1] 3. 细胞活力实验:将HEK293-hH3R细胞和SH-SY5Y细胞以5×10³个细胞/孔接种于96孔板,SB-649868(0.1 nM–10 μM)处理72小时;加入0.5 mg/mL MTT试剂孵育4小时,DMSO溶解甲臜结晶后检测570 nm吸光度,评估细胞活力[1] |
| 动物实验 |
1. 小鼠睡眠-觉醒周期评估方案:雄性 CD-1 小鼠(20-25 g)在异氟烷麻醉下接受脑电图/肌电图电极植入术,用于睡眠记录。经过 7 天的恢复期后,将小鼠随机分为两组,分别在光照期开始时(时间信号 0)通过灌胃给予 SB-649868(1、3、10 mg/kg)或载体(0.5% CMC-Na + 0.1% Tween 80,灌胃体积 0.2 mL/20 g)。连续记录6小时的脑电图/肌电图信号,并使用睡眠分析软件(10秒时段)离线对睡眠-觉醒阶段(清醒、非快速眼动睡眠、快速眼动睡眠)进行评分[1]
2. 大鼠日间过度嗜睡模型方案:雄性Sprague-Dawley大鼠(250-300克)使用旋转滚筒装置进行24小时睡眠剥夺。然后,大鼠分别灌胃给予SB-649868(3、10毫克/千克)或溶剂,并使用红外光束阻断检测器测量其运动活性4小时。采用莫里斯水迷宫评估认知能力:记录5次试验中寻找隐藏平台的逃避潜伏期,并进行探测试验以测量平台位置记忆[1] 3. 大鼠H3受体激动剂诱导的嗜睡逆转方案:雄性SD大鼠腹腔注射H3受体激动剂咪哌哌酯(1 mg/kg)以诱导嗜睡(运动活性降低60%)。30分钟后,大鼠口服SB-649868(1、3、10 mg/kg)或载体,并在开放式场地(40×40 cm)中测量1小时的运动活性(量化总运动距离)[1] |
| 药代性质 (ADME/PK) |
1. 口服生物利用度:在雄性 CD-1 小鼠中,口服 10 mg/kg 剂量的 SB-649868 的绝对口服生物利用度为 58% [1]
2. 血浆药代动力学:小鼠口服 10 mg/kg SB-649868 后,血浆峰浓度 (Cmax) 为 0.78 μM (Tmax = 1.5 小时),消除半衰期 (t₁/₂) 为 4.2 小时;血浆浓度-时间曲线下面积 (AUC₀–24h) 为 3.2 μg·h/mL [1] 3. 脑渗透性:SB-649868 在小鼠中表现出较高的脑渗透性,口服给药 (10 mg/kg) 1 小时后,脑/血浆比为 2.1;脑组织峰浓度 (Cmax,brain) 为 1.64 μM,远高于 hH3R Ki (1.6 nM) [1] 4. 代谢和排泄:SB-649868 在肝脏中经 CYP3A4 代谢为葡萄糖醛酸苷结合物(主要代谢物);小鼠口服给药 72 小时后,60% 的剂量经粪便排泄(45% 为代谢物,15% 为原药),30% 经尿液排泄(均为代谢物)[1] 5. 分布容积和清除率:小鼠静脉注射 SB-649868 (1 mg/kg) 后,分布容积 (Vd) 为 1.8 L/kg,血浆清除率 (CL) 为 12 mL/min/kg [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
1. 体外细胞毒性:SB-649868 (≤10 μM) 对 HEK293-hH3R 细胞、SH-SY5Y 细胞或原代大鼠皮层神经元无明显细胞毒性(MTT 法和 LDH 释放法检测细胞活力 >95%)[1]
2. 血浆蛋白结合率:SB-649868 在人血浆中的血浆蛋白结合率为 89%,在小鼠血浆中的血浆蛋白结合率为 87%(超滤法测定)[1] 3. 急性体内毒性:小鼠单次口服 SB-649868 (500 mg/kg) 7 天后未出现死亡或行为异常(例如共济失调、癫痫、嗜睡);小鼠口服 LD50 >500 mg/kg [1] 4. 慢性体内毒性:用 SB-649868 (30 mg/kg/天 口服) 治疗 28 天的大鼠体重增加正常,血清肝脏 (ALT/AST) 或肾脏 (肌酐、尿素) 功能标志物无变化;脑、肝、肾和心脏的组织病理学分析未发现异常[1] 5. 药物相互作用:SB-649868(≤10 μM)在体外不抑制或诱导人CYP450酶(CYP1A2、2C9、2C19、2D6、3A4),且在小鼠中未观察到与咖啡因或莫达非尼(一种促醒药物)的药代动力学相互作用[1] 6. 中枢神经系统安全性:SB-649868(口服剂量高达30 mg/kg)在小鼠中不诱发癫痫发作或多动症,且在连续给药28天后未观察到神经毒性迹象(例如,海马神经元丢失)[1] |
| 参考文献 | |
| 其他信息 |
SB-649868 正在进行临床试验 NCT01030939(一项在健康志愿者中研究重复给药 SB-649868 后的安全性、耐受性、药代动力学和心脏功能的研究)。
1. SB-649868 是一种强效、选择性强且能穿透血脑屏障的组胺 H3 受体反向激动剂,由葛兰素史克公司开发,作为治疗过度睡眠障碍(例如,发作性睡病、特发性嗜睡症)的先导化合物。[1] 2. SB-649868 通过作为 H3 受体的反向激动剂发挥其促醒作用:它抑制突触前 H3 自身受体的组成性活性,从而增加下丘脑(大脑的睡眠-觉醒调节中心)中的组胺释放。并增强清醒度[1] 3. 与传统的促醒药物(例如莫达非尼)不同,SB-649868 通过组胺能系统发挥作用,耐受性和滥用风险较低,啮齿动物自我给药研究已证实了这一点[1] 4. SB-649868 为 LML134 的开发提供了结构模板,LML134 是一种第二代 H3R 反向激动剂,经过优化,可改善其药代动力学并提高治疗过度睡眠障碍的临床疗效[1] 5. SB-649868 尚未获得 FDA 批准或正式的临床适应症;它是一种临床前先导化合物,其类似物 LML134 已进入治疗过度睡眠障碍的 I 期临床试验[1] |
| 分子式 |
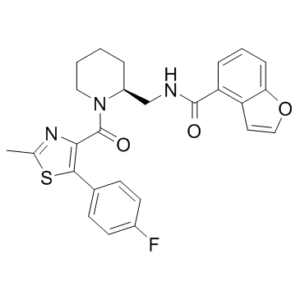
C26H24FN3O3S
|
|
|---|---|---|
| 分子量 |
477.55
|
|
| 精确质量 |
477.152
|
|
| 元素分析 |
C, 65.39; H, 5.07; F, 3.98; N, 8.80; O, 10.05; S, 6.71
|
|
| CAS号 |
380899-24-1
|
|
| 相关CAS号 |
|
|
| PubChem CID |
25195495
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| LogP |
5.941
|
|
| tPSA |
107.17
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
5
|
|
| 重原子数目 |
34
|
|
| 分子复杂度/Complexity |
734
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
O=C(C1=C(C2=CC=C(F)C=C2)SC(C)=N1)N3CCCC[C@H]3CNC(C4=C(C=CO5)C5=CC=C4)=O
|
|
| InChi Key |
ZJXIUGNEAIHSBI-IBGZPJMESA-N
|
|
| InChi Code |
InChI=1S/C26H24FN3O3S/c1-16-29-23(24(34-16)17-8-10-18(27)11-9-17)26(32)30-13-3-2-5-19(30)15-28-25(31)21-6-4-7-22-20(21)12-14-33-22/h4,6-12,14,19H,2-3,5,13,15H2,1H3,(H,28,31)/t19-/m0/s1
|
|
| 化学名 |
N-[[(2S)-1-[5-(4-fluorophenyl)-2-methyl-1,3-thiazole-4-carbonyl]piperidin-2-yl]methyl]-1-benzofuran-4-carboxamide
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.24 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.24 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0940 mL | 10.4701 mL | 20.9402 mL | |
| 5 mM | 0.4188 mL | 2.0940 mL | 4.1880 mL | |
| 10 mM | 0.2094 mL | 1.0470 mL | 2.0940 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT01030939 | Completed | Drug: SB-649868 | Sleep Disorders | GlaxoSmithKline | August 27, 2009 | Phase 1 |
| NCT01299597 | Completed | Drug: Atorvastatin Drug: Simvastatin Drug: SB649868 |
Sleep Disorders | GlaxoSmithKline | January 18, 2010 | Phase 1 |
| NCT00495729 | Completed | Drug: SB-649868 Drug: Placebo Drug: Simvastatin |
Sleep Initiation and Maintenance Disorders |
GlaxoSmithKline | April 18, 2007 | Phase 1 |
| NCT00426816 | Completed | Drug: SB-649868 Drug: Placebo |
Sleep Initiation and Maintenance Disorders |
GlaxoSmithKline | December 2006 | Phase 2 |
| NCT00520663 | Completed | Drug: 14C-SB649868 | Sleep Initiation and Maintenance Disorders |
GlaxoSmithKline | June 8, 2007 | Phase 1 |
 Orexin A-13C18,15N3 (human, rat, mouse) TFA
Orexin A-13C18,15N3 (human, rat, mouse) TFA
 OX2-2303
OX2-2303
 Orexin receptor antagonist 7
Orexin receptor antagonist 7
 Alixorexton enantiomer
Alixorexton enantiomer
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
