| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
OX1 Receptor ( IC50 = 3.76 nM ); OX2 Receptor ( IC50 = 531 nM ); OX1 Receptor ( Ki = 1.1 nM ); OX2 Receptor ( Ki = 129 nM )
SB-674042 is a selective, nonpeptide antagonist of the human orexin-1 (OX₁) receptor. Its binding affinity (Ki) was not directly provided in competition assays, but saturation and kinetic studies determined its equilibrium dissociation constant (Kd). [1] The radiolabeled form, [³H]SB-674042, binds to the human OX₁ receptor stably expressed in CHO cells with a Kd of 3.76 ± 0.45 nM in a membrane-based scintillation proximity assay (SPA) format and a Kd of 5.03 ± 0.31 nM in a whole-cell assay format. [1] In functional calcium mobilisation assays, SB-674042 acts as a competitive antagonist at the OX₁ receptor with an inhibition constant (Kb) of 1.1 ± 0.1 nM. It shows >100-fold selectivity over the human orexin-2 (OX₂) receptor (Kb at OX₂ = 129 ± 15 nM). [1] |
|---|---|
| 体外研究 (In Vitro) |
SB-674042 ([3H])(0.2-24 nM;2 h)具有高亲和力,可作为放射性配体,适合标记在 CHO 细胞中稳定表达的人 OX1 受体[1]。 SB-674042(5 μM;4 ℃ 30 分钟,37 ℃ 3 小时)可降低 CB1 受体激动剂在共表达 orexin-1 和 CB1 受体的 HEK293 细胞中磷酸化 ERK1/2 的效力[2]。 SB-674042(1 μM;24 小时)可消除 INS-1 细胞中 Orexin-A(1 nM-1 μM;24 小时)引起的 mTOR 磷酸化增加,表明由orexin-A 诱导的 mTOR 通路激活依赖于激活的 OX1 受体[3]。 Western Blot 分析[3] 细胞系:INS-1 细胞 浓度:1 μM 孵育时间:24 小时;与 1 μM Orexin-A 一起作用 24 小时 结果:降低 Orexin-A 诱导的 mTOR 磷酸化水平。
[³H]SB-674042 对表达于CHO细胞中的人OX₁受体表现出可饱和的高亲和力结合,在全细胞中特异性结合占总结合的70%以上,在膜制剂中占80%以上。在野生型CHO细胞膜中未检测到特异性结合。[1] 在使用FLIPR技术的钙动员实验中,SB-674042 能够有效且竞争性地拮抗食欲素-A(10 nM)在表达OX₁受体的CHO细胞中诱导的细胞内钙升高,Kb为 1.1 ± 0.1 nM。[1] SB-674042 对一系列其他受体(包括5-羟色胺能、多巴胺能、肾上腺素能和嘌呤能受体)在浓度高达10 µM时均未显示显著亲和力,表明其对OX₁受体具有高选择性。[1] |
| 体内研究 (In Vivo) |
SB-674042(0.3 nM/0.3 μL;icv;单剂量)可减少小鼠压力替代模型 (SMA) 中动物的情境和提示恐惧冻结反应[4]。动物模型:应激小鼠模型(雄性C57BL/6NHsd小鼠,22-26 g)[4] 剂量:0.3 nM/0.3 μL 给药方式:脑室内注射;让小鼠遭受 4 天的社会攻击(第 1-4 天) 结果:小鼠中 39.4% 的逃避表型和 60.6% 的停留表型。
|
| 酶活实验 |
[3H]SB-674042全细胞结合试验[1]
在96孔Packard培养板中培养过夜后,丢弃培养基,在25°C下将细胞在含有150 mM NaCl、20 mM HEPES和0.5%牛血清白蛋白(pH 7.4)的缓冲液中孵育60分钟。通过用不同浓度的[3H]SB-674042(0.2-24 nM)孵育细胞进行饱和研究;总测定体积为250μl。通过用0.1M NaOH裂解细胞并使用Bradford法(Bradford,1976)以牛血清白蛋白(BSA)为标准来测定蛋白质含量。 通过在加入[3H]SB-674042后1-60分钟测量[3H]SB-674042(3 nM)的特异性结合来进行关联动力学研究。对于解离研究,细胞首先与[3H]SB-674042(3 nM)孵育60分钟。然后在加入3μM SB-408124后2-120分钟测量特异性结合。通过用[3H]SB-674042(3 nM)和不同浓度的测试化合物孵育细胞进行竞争研究。用250μl冰冷的磷酸盐缓冲盐水洗涤细胞三次,终止所有测定。向每个孔中加入100μl Microscint 40,将平板在室温下放置2小时。然后使用Packard Topcount测量细胞相关放射性,计数时间为2分钟孔-1。 [3H]SB-674042基于膜的SPA结合测定[1] CHO-K1_OX1细胞膜(75μg ml−1)通过与小麦胚芽凝集素-聚乙烯基甲苯(WGA-PVT)闪烁邻近试验(SPA)珠(5 mg ml−l)在含有25 mM HEPES、2.5 mM MgCl2、0.5 mM EDTA和0.025%杆菌肽(pH 7.4)的缓冲液中在4°C下振荡1小时进行预偶联。珠膜悬浮液在300×g下离心,并重新悬浮在相同体积的室温试验缓冲液中。在96孔Packard Optiplate中,将100μl的珠膜悬浮液与[3H]SB-674042(5 nM)一起孵育,总测定体积为200μl,使最终蛋白质浓度为7.5μg孔-1。非特异性结合是在3μM SB-408124存在下剩余的结合。将分析板摇动10分钟,然后在室温下孵育4小时,然后在Packard TopCount闪烁计数器上计数(计数时间2分钟,孔-1)。 通过在[3H]SB-674042(0.1-20 nM)的浓度范围内孵育珠膜(相当于7.5μg蛋白质孔-1和2.5 mg珠ml-1)进行饱和研究。使用Bradford法(Bradford,1976)以牛血清白蛋白为标准测定蛋白质含量。通过在添加珠膜(相当于7.5μg蛋白质孔-1和2.5 mg珠ml-1)后1-30分钟测量[3H]SB-674042(5 nM)的特异性结合来进行关联动力学研究。对于解离研究,首先将珠膜与[3H]SB-674042(5 nM)一起孵育30分钟。然后在加入3μM SB-408124后2-120分钟测量特异性结合。通过用[3H]SB-674042(5 nM)和不同浓度的测试化合物孵育珠膜(相当于7.5μg蛋白质孔-1和2.5 mg珠ml-1)进行竞争研究。 |
| 细胞实验 |
细胞系:INS-1细胞
浓度:1 μM 孵育时间:24小时;与 1 μM Orexin-A 一起作用 24 小时 结果:降低 Orexin-A 诱导的 mTOR 磷酸化水平。 培养大鼠胰岛素瘤INS-1细胞,并用不同浓度的食欲素-A、有或没有OX1受体选择性拮抗剂SB-674042或磷脂酰肌醇3-激酶/mTOR拮抗剂PF-04691502处理。使用INS-1细胞进行胰岛素释放实验、蛋白质印迹分析和统计分析。 结果:我们的结果表明,用食欲素-A处理细胞会以浓度依赖的方式增加OX1受体的表达和mTOR的磷酸化。用食欲素-A处理的细胞也观察到胰岛素分泌的增加。我们通过使用OX1受体选择性拮抗剂SB-674042或磷脂酰肌醇3-激酶/mTOR拮抗剂PF-04691502进一步证明,胰岛素分泌的减少取决于OX1受体和mTOR信号通路的激活,这消除了食欲素-A治疗的影响。 结论:我们的研究结果表明,食欲素-A/OX1受体通过激活AKT及其下游靶点mTOR来刺激胰岛素分泌。因此,食欲素可能在mTOR的参与下调节细胞存活的能量平衡[3]。 全细胞放射性配体结合实验: 将稳定表达人OX₁受体的CHO-K1细胞接种于96孔板。进行饱和实验时,将细胞与一系列浓度(0.2-24 nM)的[³H]SB-674042在测定缓冲液中孵育;进行竞争实验时,使用固定浓度(3 nM)。非特异性结合在3 µM SB-408124存在下定义。孵育后,用冰预冷的缓冲液洗涤细胞,添加闪烁液,并使用微板闪烁计数器定量细胞相关的放射性。[1] 基于膜的SPA结合实验: 将来自CHO-K1_OX₁细胞的膜与小麦胚芽凝集素包被的闪烁亲近测定(SPA)珠预偶联。将珠-膜复合物与[³H]SB-674042(饱和实验0.1-20 nM;竞争实验5 nM)在测定缓冲液中孵育。非特异性结合用3 µM SB-408124确定。振荡孔板,孵育,然后在闪烁计数器上计数。[1] 钙动员实验(FLIPR): 将稳定表达人OX₁或OX₂受体的CHO-DG44细胞接种于96孔板,并用钙敏感荧光染料Fluo-3 AM加载。洗涤细胞后,与或不与SB-674042(或其他拮抗剂)孵育30分钟。使用荧光成像板读数仪(FLIPR)监测添加10 nM食欲素-A前后的荧光信号。根据对食欲素-A诱导的钙反应的抑制曲线计算拮抗剂的效价(Kb)。[1] |
| 动物实验 |
应激诱导小鼠模型(雄性C57BL/6NHsd小鼠,22-26 g)
0.3 nM/0.3 μL 脑室内注射;小鼠接受4天的社交攻击(第1-4天) 这些实验的主要处理方法是通过拮抗剂SB-674042抑制BLA Orx1R(0.3 nmol/0.3 μL,在第3天互动前1小时双侧BLA内注射),并与Orx1R刺激(通过OrxA + Orx2R拮抗剂实现)或短发夹基因敲低(在SAM互动前30天开始双侧BLA内转染)进行对比。考虑到给药时间的不同,这些处理方法根据预先设定的假设分别进行和分析。所有行为学测量均在动物活跃的暗周期进行,包括逃避行为(使用顶端隧道)、停留行为(在有陌生攻击者的SAM实验区内停留)、对逃生孔的注意力持续时间、逃避潜伏期(针对逃避组小鼠)、恐惧条件性冻结反应(在社交互动非条件刺激[US]出现之前,对音调[CS]和环境刺激的反应,以及在没有US的情况下作为条件反应[第5天的CR]进行测量)和食物摄入量。因此,处理组包括笼养对照组,以及对逃避组和停留组小鼠进行BLA内注射SB-674042(或载体、OrxA、OrxA + MK-1064、MK-1064)。此外,转基因处理组包括笼养对照组、BLA内注射AAV-Orx1R-shRNA组和BLA内注射AAV-scramble-shRNA组。采集脑组织和血液样本,利用RNAscope技术对基底外侧杏仁核(BLA)中HCRTR1、HCRTR2、钙结合蛋白(CALB1)、钙/钙调蛋白激酶2α(CAMKIIα)、谷氨酸脱羧酶(GAD1)和小白蛋白(PVALB)的基因表达进行可视化分析,并采用酶联免疫吸附试验(ELISA)测定血浆中应激激素皮质酮的浓度。此外,还采用RT-qPCR技术检测BLA组织中HCRTR1、HCRTR2、PLCB1、MAPK1、MAPK3、BDNF和GAPDH(管家基因)的基因表达。所有实验设计和统计分析均基于预先设定的假设,采用双因素重复测量方差分析、双因素方差分析、单因素方差分析、回归分析和t检验,并在适当情况下进行事后分析。[4] |
| 参考文献 |
|
| 其他信息 |
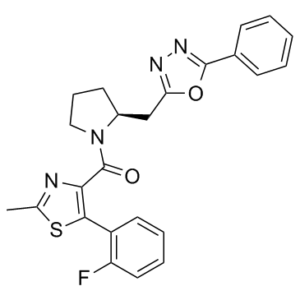
1. 本研究表征了一种新型非肽类拮抗剂放射性配体[(3)H]SB-674042(1-(5-(2-氟苯基)-2-甲基噻唑-4-基)-1-((S)-2-(5-苯基-(1,3,4)恶二唑-2-基甲基)-吡咯烷-1-基)-甲酮)与稳定表达于中国仓鼠卵巢 (CHO) 细胞中的人食欲素-1 (OX(1)) 受体的结合情况。研究采用了全细胞测定和基于细胞膜的闪烁邻近测定 (SPA) 两种方法。2. [(3)H]SB-674042 的特异性结合在全细胞和细胞膜测定中均呈饱和状态。分析表明存在一个高亲和力结合位点,在全细胞和膜结合状态下,其Kd值分别为3.76±0.45 nM和5.03±0.31 nM,相应的Bmax值分别为30.8±1.8 pmol/mg蛋白和34.4±2.0 pmol/mg蛋白。动力学研究也得到了类似的Kd值。3. 全细胞竞争实验表明,天然食欲素肽对OX1受体的亲和力较低,其中食欲素A的亲和力约为食欲素B的五倍(Ki值分别为318±158 nM和1516±597 nM)。 4. SB-334867、SB-408124(1-(6,8-二氟-2-甲基-喹啉-4-基)-3-(4-二甲氨基苯基)-脲)和SB-410220(1-(5,8-二氟-喹啉-4-基)-3-(4-二甲氨基苯基)-脲)在全细胞(K(i) 值分别为 99±18、57±8.3 和 19±4.5 nm)和膜(K(i) 值分别为 38±3.6、27±4.1 和 4.5±0.2 nm)形式中均对 OX(1) 受体表现出高亲和力。 5. 钙动员研究表明,SB-334867、SB-408124 和 SB-410220 均为 OX(1) 受体的功能性拮抗剂,其效力与其在放射性配体结合试验中测得的亲和力相符,且对食欲素-2 受体的选择性约为 50 倍。6. 这些研究表明,[(3)H]SB-674042 是 OX(1) 受体的特异性高亲和力放射性配体。这种放射性配体的出现将成为研究OX(1)受体生理功能的宝贵工具。[1]在HEK293细胞中诱导表达后,人食欲素-1受体被靶向定位于细胞表面,但在暴露于肽类激动剂食欲素A后发生内吞作用。相比之下,人大麻素CB1受体的组成型表达主要导致其在细胞内呈点状分布,这与自发性、非激动剂依赖性内吞作用一致。在CB1受体存在的情况下表达食欲素-1受体,导致两种受体均表现出自发性内吞表型。单细胞荧光共振能量转移成像显示,这两种受体以异二聚体/寡聚体的形式存在于细胞内囊泡中。向仅表达CB1受体的细胞中添加CB1受体拮抗剂SR-141716A,导致受体重新定位至细胞表面。尽管SR-141716A对食欲素-1受体没有显著的亲和力,但在共表达CB1受体的细胞中,SR-141716A处理也能使食欲素-1受体重新定位到细胞表面。用食欲素-1受体拮抗剂SB-674042处理共表达食欲素-1和CB1受体的细胞,也导致两种受体重新定位到细胞表面。SR-141716A处理仅在共表达这两种受体的细胞中降低了食欲素A激活丝裂原活化蛋白激酶ERK1/2的效力。SB-674042处理也仅在共表达这两种受体时降低了CB1受体激动剂磷酸化ERK1/2的效力。这些研究引入了一种全新的药理学范式,即配体通过调节受体异二聚体,从而调控与其本身没有显著亲和力的受体的功能。[2]
背景:应激通过对特定神经环路元件进行选择性分子修饰,产生不同的行为反应。食欲素(Orx)系统靶向基底外侧杏仁核(BLA)中该神经环路的关键组成部分。方法:我们评估了BLA内Orx1受体(Orx1Rs)在小鼠应激诱导表型表达中的作用。我们利用应激替代模型(一种可产生两种行为表型的社会应激范式),采用急性药理学抑制(SB-674042)和基因敲低(AAV-U6-Orx1R-shRNA)策略,对基底外侧杏仁核(BLA)内Orx1R的作用进行了表征。结果:在BLA中,我们观察到Orx1R(Hcrtr1)mRNA主要在CamKIIα+谷氨酸能神经元中表达,而在GABA能(γ-氨基丁酸能)细胞中表达较少。虽然BLA中Hcrtr1和Orx2受体(Hcrtr2)mRNA的表达存在轻微重叠,但我们发现这些受体通常在不同的细胞中表达。在表型形成后,拮抗基底外侧杏仁核(BLA)内的Orx1R可使行为表达从应激敏感型(停留)转变为应激耐受型(逃避),这种效应可通过基因敲低模拟。急性抑制BLA中的Orx1R也降低了停留型动物的情境恐惧和线索恐惧冻结反应。这种表型特异性的行为改变伴随着偏向性的分子转录,即Hcrtr2优于Hcrtr1,Mapk3优于Plcb1,以及Bdnf mRNA水平升高。结论:拮抗Orx1R可导致BLA内基因表达的功能重组,从而促进Hcrtr2、Mapk3和Bdnf表达的升高。这些结果共同为基底外侧杏仁核(BLA)内平衡促应激和抗应激反应的受体驱动机制提供了证据。[4] SB-674042的结构被鉴定为1-(5-(2-氟苯基)-2-甲基噻唑-4-基)-1-((S)-2-(5-苯基-(1,3,4)恶二唑-2-基甲基)-吡咯烷-1-基)-甲酮。[1] 放射性标记版本[³H]SB-674042(比活度27 Ci mmol⁻¹)被认为是首个选择性非肽类OX₁受体拮抗剂放射性配体。它是一种用于研究OX₁受体定位和功能的宝贵工具化合物。 [1] 结合动力学研究表明,[³H]SB-674042与OX₁受体的结合在30-60分钟内达到平衡,并且过量非竞争剂诱导的解离是单相的。[1] |
| 分子式 |
C24H21FN4O2S
|
|---|---|
| 分子量 |
448.51254
|
| 精确质量 |
448.137
|
| 元素分析 |
C, 64.27; H, 4.72; F, 4.24; N, 12.49; O, 7.13; S, 7.15
|
| CAS号 |
483313-22-0
|
| PubChem CID |
10204153
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
5.092
|
| tPSA |
100.36
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
32
|
| 分子复杂度/Complexity |
652
|
| 定义原子立体中心数目 |
1
|
| SMILES |
FC1=C(C2=C(C(N3[C@H](CC4=NN=C(C5=CC=CC=C5)O4)CCC3)=O)N=C(C)S2)C=CC=C1
|
| InChi Key |
HYBZWVLPALMACV-KRWDZBQOSA-N
|
| InChi Code |
InChI=1S/C24H21FN4O2S/c1-15-26-21(22(32-15)18-11-5-6-12-19(18)25)24(30)29-13-7-10-17(29)14-20-27-28-23(31-20)16-8-3-2-4-9-16/h2-6,8-9,11-12,17H,7,10,13-14H2,1H3/t17-/m0/s1
|
| 化学名 |
[5-(2-fluorophenyl)-2-methyl-1,3-thiazol-4-yl]-[(2S)-2-[(5-phenyl-1,3,4-oxadiazol-2-yl)methyl]pyrrolidin-1-yl]methanone
|
| 别名 |
SB-674042; SB 674042; SB-674042; 483313-22-0; SB 674042; SB674042; [5-(2-fluorophenyl)-2-methyl-1,3-thiazol-4-yl]-[(2S)-2-[(5-phenyl-1,3,4-oxadiazol-2-yl)methyl]pyrrolidin-1-yl]methanone; CHEMBL2110363; DTXSID90436738; (S)-(5-(2-Fluorophenyl)-2-methylthiazol-4-yl)(2-((5-phenyl-1,3,4-oxadiazol-2-yl)methyl)pyrrolidin-1-yl)methanone; SB674042
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~25 mg/mL (~55.7 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.43 mg/mL (3.19 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 14.3 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.43 mg/mL (3.19 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 14.3mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.43 mg/mL (3.19 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2296 mL | 11.1480 mL | 22.2960 mL | |
| 5 mM | 0.4459 mL | 2.2296 mL | 4.4592 mL | |
| 10 mM | 0.2230 mL | 1.1148 mL | 2.2296 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA




 463611831
463611831
