| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
Flk-1 (IC50 = 1.23 μM)
Semaxanib (SU5416) is a potent and selective inhibitor of the vascular endothelial growth factor receptor-2 (VEGFR-2/Flk-1/KDR), which inhibits tyrosine kinase activity and downstream signaling. [1] It also inhibits the stem cell factor receptor tyrosine kinase c-Kit, often expressed in acute myelogenous leukemia cells. [8] Semaxanib (SU-5416) selectively inhibits vascular endothelial growth factor receptor 2 (VEGFR2/Flk-1/KDR) tyrosine kinase (IC₅₀ = 140 nM for recombinant VEGFR2; IC₅₀ = 250 nM for VEGFR2 phosphorylation in HUVECs) [1] Semaxanib (SU-5416) shows weak inhibitory activity against PDGFRβ (IC₅₀ = 3.0 μM) and c-Kit (IC₅₀ = 5.0 μM), with no significant effect on EGFR, FGFR, or Abl (IC₅₀ > 10 μM) [1] |
|---|---|
| 体外研究 (In Vitro) |
Semaxanib 抑制过表达 Flk-1 的 NIH 3T3 细胞中 Flk-1 受体的 VEGF 依赖性磷酸化,IC50 为 1.04 μM。 Semaxanib 抑制 NIH 3T3 细胞中 PDGF 依赖性自磷酸化,IC50 为 20.3 μM。 Semaxanib 以剂量依赖性方式抑制 VEGF 和 FGF 驱动的有丝分裂,IC50 分别为 0.04 和 50 μM。 Semaxanib治疗对C6神经胶质瘤、Calu 6肺癌、A375黑色素瘤、A431表皮样癌和SF767T神经胶质瘤细胞的体外生长没有影响(所有IC50 > 20 μM)。激酶测定:将 3T3 Flk-1 细胞的溶解膜添加到已预先涂有可识别 Flk-1 的单克隆抗体的聚苯乙烯 ELISA 板上。在 4℃ 下与裂解物孵育过夜后,将 SU5416 的系列稀释液添加到免疫定位受体中。为了诱导受体的自身磷酸化,将不同浓度的 ATP 添加到含有系列稀释的 SU5416 溶液的 ELISA 板孔中。自磷酸化在室温下进行 60 分钟,然后用 EDTA 终止。通过将免疫定位受体与针对磷酸酪氨酸的生物素化单克隆抗体一起孵育来确定各个孔中 Flk-1 受体上存在的磷酸酪氨酸的量。除去未结合的抗磷酸酪氨酸抗体后,将亲和素缀合的辣根过酸酶 H 添加到孔中。将稳定形式的 3,3 9,5,5 9-四甲基联苯胺二盐酸盐和 H2O2 添加到孔中。测定的颜色读数允许显色 30 分钟,并用 H2SO4 终止反应。细胞测定:将 HUVEC 接种于 96 孔平底板(1×104 个细胞/100 μL/孔)中,加入含 0.5% 热灭活 FBS 的 F-12K 培养基中,37 ℃ 培养 24 h,使细胞静止。 。然后添加在含有 1% DMSO 的培养基中制备的化合物的连续稀释液 2 小时,然后添加促有丝分裂浓度的 VEGF(5 ng/mL 或 20 ng/mL)或酸性成纤维细胞生长因子(0.25-5 ng/mL)在媒体中。测定中 DMSO 的终浓度为 0.25%。 24小时后,添加[3H]胸苷(1μCi/孔)或BrdUrd,并将细胞单层再孵育24小时。分别使用液体闪烁计数器或 BrdUrd ELISA 对细胞摄取的 [3H] 胸苷或 BrdUrd 进行定量。
塞马西尼(SU-5416)剂量依赖性抑制血管内皮生长因子(VEGF)诱导的人脐静脉内皮细胞(HUVECs)增殖,IC₅₀为0.3μM。1μM时,可抑制HUVECs迁移约80%、管腔形成约85%,并阻断VEGF介导的VEGFR2磷酸化及下游Akt/ERK1/2信号通路[1] 塞马西尼(SU-5416)抑制血小板衍生生长因子-BB(PDGF-BB)诱导的血管平滑肌细胞(VSMCs)增殖,IC₅₀为2.5μM。1μM时可减少A549肺癌细胞中血管生成因子(VEGF、bFGF)的分泌约40%[2] 在黑色素瘤细胞系(A375、SK-MEL-28)中,塞马西尼(SU-5416)在浓度高达10μM时无直接抗增殖作用,但与沙利度胺联合使用时,可增强其细胞毒性约30%[5] |
| 体内研究 (In Vivo) |
Semaxanib 剂量相关地抑制体内 A375 肿瘤的生长。每日腹膜内施用 Semaxanib 的 DMSO 中的 SU5416 时,观察到皮下肿瘤生长抑制 >85%,且没有可测量的毒性。 Semaxanib 显示出广谱抗肿瘤活性。 SU5416 显着抑制所测试的 10 个肿瘤系中的 8 个(A431、Calu-6、C6、LNCAP、EPH4-VEGF、3T3HER2、488G2M2 和 SF763T 细胞)的皮下生长,平均死亡率为 2.5%。 Semaxanib(25 mg/kg/天)显示出有效的抗血管生成活性,导致肿瘤微血管系统的总血管密度和功能血管密度显着降低。
Semaxanib/SU5416 +缺氧(SuHx)大鼠模型是重度肺动脉高压常用模型。虽然已知缺氧暴露可以被另一种类型的打击(例如,卵清蛋白敏化)所取代,但尚不清楚肺血流量异常(PBF)是否可以取代缺氧暴露,这种异常一直被认为会引起肺血管的病理改变。本研究研究 Semaxanib/SU5416联合肺切除术(PNx)诱导对侧肺PBF异常是否足以诱导大鼠严重肺动脉高压(PAH)。Sprague Dawley大鼠采用SuPNx方案(SU5416 +联合左侧全肺切除术)或标准SuHx方案,并在启动后第2周和第6周进行模型比较。SuHx和SuPNx模型均表现出广泛的闭塞性血管重构,导致第6周右心室收缩压升高。两种模型的肺血管中均观察到类似的炎症反应,内皮细胞增殖和凋亡增加。本研究描述了SuPNx模型,该模型在6周时具有严重的多环芳烃,可以作为SuHx模型的替代方案。我们的研究以及先前对肺动脉高压实验模型的研究表明,PAH的典型组织病理学表现,包括闭塞性病变、炎症、细胞更新增加和持续的细胞凋亡,代表了一种疾病的最终共同途径,这种疾病可以作为肺脉管系统受到各种损害的结果而发展。[3] 最近报道了肺动脉高压(PAH)期间肺血管和右心室细胞显著的糖酵解移位。由于该疾病的任何变体的晚期并发症和破坏性过程,因此非常需要重现PAH潜在代谢重编程的动物模型。我们的目的是通过代谢组学分析和分子成像原位研究PAH小鼠模型肺和右心室的代谢重编程。PAH是由慢性缺氧暴露加血管内皮生长因子受体抑制剂Semaxanib/SU5416)治疗引起的。采用磁共振波谱法对肺和右心室样本进行分析。采用正电子发射断层扫描研究体内能量代谢。我们的研究结果表明,肺样本的代谢组学分析清楚地确定了糖酵解途径的显著改变。我们还证实了谷氨酰胺代谢的上调和脂质代谢的改变。此外,我们还发现了肺组织中甘氨酸和胆碱代谢的改变。在右心室样本中也证实了代谢重编程。乳酸和丙氨酸,糖酵解氧化的终点,在多环芳烃小鼠中发现浓度增加。谷氨酰胺和牛磺酸浓度与特定心室肥厚特征相关。我们证明了人类PAH的大多数代谢特征在缺氧加Semaxanib/SU5416小鼠模型中被检测到,它可能成为测试这种严重疾病的新靶向治疗的有价值的工具。[4] 塞马西尼(SU-5416)抑制裸鼠体内多种异种移植瘤的生长和血管生成,包括结直肠癌(Colo205)、乳腺癌(MDA-MB-231)和肺癌(A549)。腹腔注射20mg/kg/天,持续21天,肿瘤体积减少55-70%,瘤内微血管密度(CD31阳性)降低60-75%[1] 通过活体多荧光视频显微镜观察,塞马西尼(SU-5416)(10mg/kg/天,腹腔注射,持续14天)可破坏Lewis肺癌异种移植瘤的微循环,降低血管通透性并诱导血管退化[2] 塞马西尼(SU-5416)(20mg/kg/周,皮下注射)与肺切除术联合使用,可诱导大鼠发生严重肺动脉高压,平均肺动脉压较假手术对照组升高2.5倍[3] 在转移性黑色素瘤患者的II期临床研究中,塞马西尼(SU-5416)(65mg/m²,每周两次,静脉注射)联合沙利度胺的疾病控制率(稳定疾病)为28%,无部分缓解或完全缓解病例[5] 在晚期实体瘤患者的I期研究中,塞马西尼(SU-5416)(40-80mg/m²,每周一次,静脉注射)联合顺铂和伊立替康的毒性可控,但抗肿瘤活性有限,15%的患者达到稳定疾病[6] |
| 酶活实验 |
Semaxanib (SU5416) 通过竞争性结合ATP位点抑制VEGFR-2酪氨酸激酶活性,激酶抑制实验显示其对VEGFR-2具有高度选择性。
[1]
然后用 3T3 Flk-1 细胞的可溶性膜填充预涂有 Flk-1 特异性单克隆抗体的聚苯乙烯 ELISA 板。在 4°C 下与裂解物一起孵育过夜后,将 SU5416 的系列稀释液添加到免疫定位受体中。含有连续稀释的 SU5416 溶液的 ELISA 板孔中充满了不同浓度的 ATP,以引起受体的自身磷酸化。室温下 60 分钟后,使用 EDTA 终止自磷酸化过程。将免疫定位受体与针对磷酸酪氨酸的生物素化单克隆抗体一起孵育,以测量每个孔中 Flk-1 受体上磷酸酪氨酸的量。提取未结合的抗磷酸酪氨酸抗体后,将与亲和素缀合的智人添加到孔中。将稳定形式的三、三、五、九四甲基联苯胺二盐酸盐与H2O2一起添加到每个孔中。 H2SO4 用于在 30 分钟后停止反应,让测定的颜色读数继续进行。 将重组人VEGFR2(Flk-1)激酶结构域与ATP及特异性多肽底物在系列稀释的塞马西尼(SU-5416)存在下孵育,反应在37°C下进行60分钟,采用放射免疫法检测磷酸化底物。通过与溶媒对照组的放射性对比计算抑制率,从量效曲线中得出IC₅₀值[1] 采用相同方案检测塞马西尼(SU-5416)对重组PDGFRβ、c-Kit、EGFR和FGFR激酶的抑制活性,反应条件保持一致,通过确定IC₅₀值证实其对VEGFR2的选择性靶向[1] |
| 细胞实验 |
在内皮细胞培养实验中,Semaxanib (SU5416) 可抑制VEGF诱导的细胞增殖(BrdU掺入法)和迁移(Transwell实验)。Western blot分析证实其能有效抑制VEGFR-2磷酸化。
[1]
在HepG2和TAMH细胞中,细胞毒性实验显示其具有剂量依赖性的抑制活性,IC50值在微摩尔级别。 [8] 将 HUVEC 接种到 96 孔平底板(1×10 4 细胞/100 μL/孔)中,并在含有以下成分的 F-12K 培养基中于 37 °C 培养 24 小时以使细胞静止0.5% 热灭活胎牛血清。添加在含有 1% DMSO 的培养基中制备的化合物的连续稀释液两小时后,然后向培养基中补充促有丝分裂浓度的酸性成纤维细胞生长因子 (0.5–5 ng/mL) 或 VEGF(5 ng/mL 或 20 ng) /毫升)。在测定中,最终DMSO浓度为0.25%。一整天后,添加 BrdUrd 或 [ 3 H]胸苷(1 μCi/孔),将单层细胞再培养 24 小时。可以使用液体闪烁计数器或 BrdUrd ELISA 分别定量细胞摄取 [ 3 H] 胸苷或 BrdUrd。 将HUVECs以5×10³个细胞/孔接种到96孔板中,血清饥饿12小时。加入塞马西尼(SU-5416)(0.05-5μM)预处理1小时后,用VEGF(50ng/mL)刺激细胞。72小时后,采用四唑盐法检测细胞活性并计算IC₅₀值。蛋白质印迹分析中,用0.5-2μM药物和VEGF处理HUVECs,裂解后与抗磷酸化VEGFR2、Akt、ERK1/2和GAPDH的抗体孵育[1] 将VSMCs接种到96孔板中,用塞马西尼(SU-5416)(0.5-10μM)预处理1小时后,用PDGF-BB(20ng/mL)刺激细胞。48小时后,通过BrdU掺入法评估细胞增殖。用1μM药物处理A549细胞24小时,采用酶联免疫吸附试验(ELISA)测量VEGF/bFGF的分泌量[2] 用塞马西尼(SU-5416)(0.1-10μM)单独或与沙利度胺(10μM)联合处理A375和SK-MEL-28黑色素瘤细胞72小时,采用MTT法检测细胞活性以评估协同细胞毒性[5] |
| 动物实验 |
小鼠:本研究使用12周龄、体重20-22克的雌性BALB/c裸鼠。手术过程中采用无菌技术。在结肠上方腹壁正中做一个1厘米的小切口。将27号针头插入结肠浆膜下,植入C6细胞(0.5×10⁶个细胞/只)。植入后,将所有暴露的肠段重新塞回腹腔。使用6.0号缝线和伤口夹封闭腹膜和皮肤。术后7天取出伤口夹。从植入后第1天开始,每天一次腹腔注射50微升DMSO或塞马沙尼(SU5416)。植入后13-16天,将动物安乐死,并使用游标卡尺或称重法测量结肠局部肿瘤的生长量。肿瘤体积的计算公式为:长×宽×高。
大鼠:将60只雄性Sprague Dawley大鼠(n = 60,6-8周龄)随机分为五组:对照组(Con)、肺切除组(PNx)、塞马沙尼组(SU)、塞马沙尼+低氧组(SuHx)和塞马沙尼+肺切除组(SuPNx)。SuHx方案如下:简而言之,动物注射25 mg/kg溶于羧甲基纤维素(CMC)的塞马沙尼,然后在10%低氧条件下暴露4周,之后恢复至常氧环境。PNx组的动物接受了左侧肺切除术。在PNx手术后两天,给予25 mg/kg的semaxinib注射。对照组(Con)仅接受溶剂CMC。在基线(缺氧/手术前)、第2周和第6周,使用超声心动图评估心脏的形态和功能。将动物安乐死,并在手术或缺氧后2周和6周,通过导管测量其左心室(LV)和右心室(RV)压力。 药代动力学[5] Semaxanib血浆和尿液采样[5] 在第一个和第二个疗程的第1天采集semaxanib的血样。在给药前以及semaxanib输注后10、35和45分钟、1、1.5、2、4、6、8和24小时,用肝素化试管采集血样(5 ml)。在第一疗程的第一周,分别于 0–8 小时、8–24 小时、24–48 小时和 48–72 小时收集总尿量。收集后立即将所有样本以 3000 rpm 离心 15 分钟,转移至标记的冻存管中,并在 -80°C 下冷冻保存直至分析。 塞马沙尼的药代动力学分析[5] 使用 WinNonLin® 软件进行非房室模型建模和参数估计。采用线性梯形法计算从零时点到最后一个可定量样本采集时间点的浓度-时间曲线下面积 (AUC0–Tf)。通过将最后一次测量的浓度除以末端速率常数 (k) 来外推 AUC 至无穷大 (AUC0–inf),末端速率常数 (k) 通过线性回归计算血浆浓度-时间曲线对数线性末端部分的斜率。末端半衰期 (t½) 计算公式为 0.693/k。通过观察浓度-时间曲线确定观察到的最大血浆浓度 (Cmax) 和达峰时间 (Tmax)。将大鼠随机分为五组:对照组 (Con)、塞马沙尼/SU5416 组 (SU)、肺切除组 (PNx)、SU5416 + 低氧组 (SuHx) 和 SU5416 + 肺切除组 (SuPNx)(图 1)。SuHx 方案如前所述 (5)。简而言之,将溶于羧甲基纤维素 (CMC) 的 SU5416 (25 mg/kg) 注射到动物体内,并在低氧 (10%) 环境下暴露 4 周,之后再恢复到常氧环境。PNx 组动物接受左侧肺切除术。 PNx手术后两天,注射SU5416(25 mg/kg)。对照组仅注射溶剂CMC。在基线(缺氧/术前)、术后第2周和第6周进行超声心动图检查,以确定心脏形态和功能。术后/缺氧后2周和6周,对动物进行麻醉,并通过导管插入术测量右心室(RV)和左心室(LV)压力。动物经放血处死,称量器官重量并进行分析。[3]本研究采用已建立的小鼠肺动脉高压(PAH)模型,该模型由缺氧暴露联合塞马沙尼(SU5416)给药(HPX+SU模型)建立。健康常氧小鼠(NMX组)和健康缺氧小鼠(HPX组)作为对照。HPX+SU小鼠PAH模型已在之前的研究中得到充分表征。简而言之,将10周龄雄性C57BL/6小鼠(查尔斯河实验室)置于常压低氧(10%氧气)环境中3周(n = 25),每周仅取出一次进行皮下注射VEGF抑制剂Semaxanib/SU5416。SU5416悬浮于羧甲基纤维素(CMC)溶液中(0.5% [w/v] CMC钠、0.9% [w/v] 氯化钠、0.4% [v/v] 聚山梨醇酯80、0.9% [v/v] 苯甲醇,溶于去离子水),注射剂量为20 mg/kg。HPX小鼠(n = 12)暴露于相同的低氧条件下,并每周进行一次假注射。 NMX小鼠(n = 25)饲养于氧气水平正常的房间内。[4] 携带Colo205、MDA-MB-231或A549异种移植瘤(100-150 mm³)的裸鼠被随机分为对照组和治疗组。塞马沙尼(SU-5416)溶于DMSO,并用生理盐水稀释(最终DMSO浓度≤5%),然后以20 mg/kg/天的剂量腹腔注射,连续21天。每3天测量一次肿瘤体积,并处死小鼠收集肿瘤组织进行CD31免疫染色。[1] 将Lewis肺癌细胞植入C57BL/6小鼠体内。当肿瘤体积达到50 mm³时,小鼠接受塞马沙尼(SU-5416)(10 mg/kg/天,腹腔注射)治疗,连续14天。采用活体多荧光显微视频技术观察第 7 天和第 14 天的肿瘤微循环[2] 雄性 Sprague-Dawley 大鼠接受左侧全肺切除术,随后皮下注射 Semaxanib (SU-5416),剂量为 20 mg/kg/周,持续 3 周。通过导管测量平均肺动脉压,并收集肺组织进行血管重塑的组织病理学分析[3] |
| 药代性质 (ADME/PK) |
药代动力学和药效学[5]
所有12例患者均在第1疗程进行了塞马沙尼血浆采样,其中7例患者在第2疗程进行了采样。塞马沙尼的主要药代动力学参数汇总于表5。第1疗程的Cmax范围为1.2至3.8 μg/ml,第2疗程为1.1至3.9 μg/ml;t1/2为1.3 (± 0.31) h。第1疗程和第2疗程的药代动力学参数相似,表明未发生明显的药物蓄积或药物相互作用。此外,暴露量的药代动力学参数与单药II期研究中观察到的相似,进一步支持了无药物相互作用的结论。图1和图2分别展示了在第1疗程和第2疗程进行药代动力学采样的7例患者的塞马沙尼Cmax和AUC值的散点图。 1和2. 比较第1周期和第2周期的Cmax和AUC值,结果显示Cmax随时间推移无显著差异,但第2周期的AUC值有所下降(但无统计学意义)。药代动力学结果表明,塞马沙尼的暴露参数与单药I期研究中观察到的参数相当,提示塞马沙尼与沙利度胺之间不存在显著的药物相互作用。比较第1周期和第2周期的Cmax和AUC值,结果显示Cmax无显著差异,但第2周期的AUC值有所下降。这与其他研究结果一致,这些研究报告称,塞马沙尼在每日或每两周给药方案下,清除率可提高50-60%。清除率提高的机制尚不清楚,但可能继发于药物或预用药所需的皮质类固醇诱导肝酶活性。药效学分析显示,接受超过四个周期治疗的患者血清VEGF水平有升高的趋势。然而,由于样本量较小,无法进行统计学比较。这一观察结果与近期研究结果相符,这些研究表明尿液和血浆中VEGF水平升高可能与VEGFR抑制剂的临床疗效相关。对此的一种可能解释是,VEGFR抑制剂治疗期间,受体结合后内化的VEGF减少,从而导致循环VEGF水平升高。有限的数据表明,塞马沙尼(SU5416)口服生物利用度较差,在临床前研究中通常采用静脉或皮下注射给药。其血浆蛋白结合率较高,但因蛋白置换而导致的临床药物相互作用的可能性不大。 [9] 在晚期实体瘤患者中,静脉注射塞马沙尼(SU-5416)(65 mg/m²,每周两次)显示平均血浆半衰期为 2.8 小时,Cmax 为 1.2 μg/mL,AUC₀-∞ 为 3.6 μg·h/mL。该药物主要通过细胞色素 P450 3A4 代谢,72 小时内 70% 的剂量经粪便排出,25% 经尿液排出。[5] 在大鼠中,单次静脉注射塞马沙尼(SU-5416)(20 mg/kg)导致分布容积为 1.8 L/kg,总清除率为 0.5 L/h/kg。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
塞马沙尼的非血液学毒性[5]
该方案的主要非血液学毒性包括8例患者出现头痛,其中3例达到3-4级;以及3例患者出现3-4级血栓形成。出现严重头痛的患者均为女性,年龄43-59岁,有偏头痛史(2例)或焦虑症史(1例)。这种副作用主要发生在首次输注塞马沙尼后,并在后续输注前使用非甾体类抗炎药进行预处理后,减轻至1级或2级。然而,1例患者在第一个疗程中出现3级头痛后决定撤回知情同意。 1例患者在第一个疗程的第20天发生肺栓塞。另有2例患者分别发生3级颈内静脉血栓形成和3级锁骨下静脉血栓形成。这两例患者的螺旋CT扫描均未发现下腔静脉受累或肺栓塞迹象。这些血栓栓塞事件被认为与药物相关,受影响的患者根据方案要求退出研究。8例患者出现感觉神经病变,表现为间歇性麻木和刺痛感,主要累及上下肢,但程度通常为轻度至中度:5例为1级,2例为2级。仅1例患者在第一个疗程后出现3级感觉神经病变。该患者还出现其他神经系统症状,包括头痛、眩晕、平衡障碍,并被诊断为脑转移。下肢水肿是一种常见但可耐受的副作用(2例为1级,3例为2级)。水肿的严重程度似乎随着治疗疗程的增加而加重。其他毒性反应为轻度至中度(1级或2级),包括:乏力(11例)、便秘(3例)、高胆固醇血症(1例)和高血糖(2例)。1例患者在4个疗程后出现无症状的4级高甘油三酯血症,接受阿托伐他汀钙治疗后甘油三酯水平显著降低,得以继续以较低剂量参与研究。2例患者出现无症状的4级高血糖,与塞玛沙尼治疗前所需的糖皮质激素治疗有关。值得注意的是,治疗期间血清皮质醇水平或凝血功能检查未见显著变化;然而,仅有6例患者按照方案完成了这些实验室检查。主要非血液学毒性反应总结于表4。 塞玛沙尼的血液学毒性[5] 血液学毒性轻微,包括3例患者出现1级贫血。未观察到中性粒细胞减少症、淋巴细胞减少症或血小板减少症。我们证实,VEGFR-2酪氨酸激酶抑制剂塞马沙尼/SU5416(65 mg/m²,每周两次)联合每周一次的顺铂30 mg/m²和伊立替康50 mg/m²(第1-4周,每6周为一个疗程)具有良好的耐受性。总体而言,毒性可逆、可控,且与先前报道的SU5416毒性特征一致。在DL2剂量下,我们观察到血液学毒性高于每周一次顺铂和伊立替康的预期水平,提示更高剂量的SU5416可能会加剧该方案的血液学毒性。本研究未进行药代动力学分析,这限制了我们对该发现的全面解释,我们也不能排除药代动力学相互作用导致DL2剂量下毒性增加或重叠的可能性。我们的研究人群既往接受过大量治疗,超过四分之三的患者接受过两线或两线以上的既往治疗,这可能限制了患者对研究性治疗方案的耐受性。 尽管给予了低剂量抗血栓预防,我们观察到治疗相关的静脉血栓栓塞事件发生率高于单独使用Semaxanib/SU5416的预期发生率(0-16%)。顺铂和伊立替康均与静脉血栓栓塞相关,这些药物与SU5416联合使用可能导致了本研究中观察到的静脉血栓栓塞事件的发生。有研究假设,SU5416 与诱导血小板减少症的药物联合使用,可能由于对血小板和内皮细胞的联合作用而导致血栓栓塞事件增加,这或许也能解释我们的研究结果。[6] 在临床试验中,塞马沙尼 (SU5416) 的耐受性总体良好,副作用包括恶心、呕吐和轻度血液毒性。动物研究中未报告严重的器官毒性。 [5][6] 接受Semaxanib (SU-5416)(20 mg/kg/周,皮下注射)治疗的大鼠,在接受肺切除术后出现严重的肺动脉高压,其特征是肺小动脉中层肥厚和右心室肥厚[3] 在 II 期临床试验中,Semaxanib (SU-5416)的常见不良事件包括疲劳(62%)、恶心(58%)、呕吐(45%)、腹泻(38%)和高血压(30%)。 3/4级毒性反应包括中性粒细胞减少症(15%)、血小板减少症(10%)和可逆性肝酶升高(8%)[5] 通过平衡透析法测定,塞马沙尼(SU-5416)在人血浆中的血浆蛋白结合率约为95%[6] |
| 参考文献 |
|
| 其他信息 |
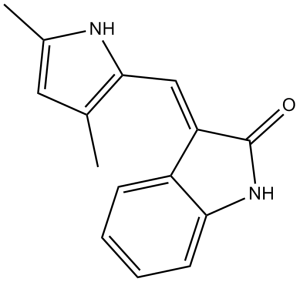
塞马沙尼是一种吲哚酮类化合物,其结构为3-亚甲基吲哚酮,其中一个亚甲基上的氢原子被3,5-二甲基吡咯-2-基取代。它具有抗肿瘤活性,可作为血管内皮生长因子受体拮抗剂、EC 2.7.10.1(受体蛋白酪氨酸激酶)抑制剂和血管生成调节剂。它属于吡咯类、吲哚酮类和烯烃类化合物。其功能与3-亚甲基吲哚酮相关。
塞马沙尼是一种喹诺酮衍生物,具有潜在的抗肿瘤活性。塞马沙尼可逆性地抑制ATP与血管内皮生长因子受体2 (VEGFR2) 酪氨酸激酶结构域的结合,从而抑制VEGF刺激的内皮细胞迁移和增殖,并减少肿瘤微血管。该药物还能抑制干细胞因子受体酪氨酸激酶c-kit的磷酸化,c-kit通常在急性髓系白血病细胞中表达。 药物适应症 已在结直肠癌和肺癌的治疗中进行研究。 SU5416是一种新型合成化合物,是Flk-1/KDR受体酪氨酸激酶的强效选择性抑制剂,目前正在进行I期临床试验,用于治疗人类癌症。体外实验表明,SU5416可抑制人内皮细胞的血管内皮生长因子依赖性有丝分裂,而不抑制多种肿瘤细胞的生长。相反,在小鼠体内以非毒性剂量全身性给予SU5416可抑制源自不同组织来源的细胞皮下肿瘤生长。SU5416的抗肿瘤作用伴随着从药物治疗动物体内切除的淡白色肿瘤的出现,这支持了该药物的抗血管生成特性。这些发现表明,药理学抑制血管内皮生长因子受体的酶活性代表了一种限制多种肿瘤类型生长的新策略。[1] 血管内皮生长因子(VEGF)在介导肿瘤血管生成和肿瘤生长中起着至关重要的作用。本文利用活体多荧光显微视频技术,研究了一种新型小分子VEGF Flk-1介导信号转导通路抑制剂SU5416对实验性胶质母细胞瘤的肿瘤血管生成和微血流动力学的直接影响。SU5416治疗显著抑制了肿瘤生长。同时,Semaxanib/SU5416 表现出强效的抗血管生成活性,导致肿瘤微血管的总血管密度和功能性血管密度均显著降低,表明治疗后肿瘤的血管生成受损,灌注严重不足。这种灌注不足并未通过血管直径的变化或无灌注血管的募集得到补偿。对肿瘤微循环的分析显示,血管生成阻断后,残余肿瘤血管中出现了显著的微血流动力学变化,例如红细胞速度和血流量较对照组更高。我们的结果表明,通过小分子抑制剂靶向 Flk-1/KDR 酪氨酸激酶的新型抗血管生成策略,是控制血管生成依赖性肿瘤生长和进展的有效方法。本研究深入探讨了体内靶向 Flk-1/KDR 对微血管的影响,并可能对未来血管生成依赖性肿瘤的治疗具有重要意义。[2] SU5416/塞马沙尼布+缺氧(SuHx)大鼠模型是常用的严重肺动脉高压模型。虽然已知缺氧暴露可被其他类型的刺激(例如卵清蛋白致敏)替代,但尚不清楚异常肺血流(PBF)是否可以替代缺氧暴露。长期以来,异常肺血流已被证实可诱发肺血管病理改变。本研究旨在探讨SU5416给药联合肺切除术(PNx)诱导对侧肺异常PBF是否足以诱导大鼠发生严重肺动脉高压(PAH)。Sprague Dawley大鼠分别接受SuPNx方案(SU5416联合左侧肺切除术)或标准SuHx方案,并在治疗后第2周和第6周对两种模型进行比较。 SuHx 和 SuPNx 模型均表现出广泛的血管闭塞性重塑,导致第 6 周右心室收缩压升高。两种模型肺血管均出现类似的炎症反应,并伴有内皮细胞增殖和凋亡增加。本研究描述了 SuPNx 模型,该模型在第 6 周表现出严重的肺动脉高压 (PAH),可作为 SuHx 模型的替代模型。我们的研究结合既往关于肺动脉高压实验模型的研究表明,PAH 的典型组织病理学表现,包括闭塞性病变、炎症、细胞周转率增加和持续性凋亡,代表了该疾病的最终共同通路,而该疾病的发生发展可能由多种肺血管损伤因素引起。 [3] 目的:本II期研究评估了小分子血管内皮生长因子(VEGF)受体2酪氨酸激酶抑制剂塞玛沙尼(Semaxanib)与沙利度胺联合治疗转移性黑色素瘤患者的疗效、耐受性、药代动力学(PK)和药效学(PD)特征。 患者和方法:既往接受过至少一种生物制剂和/或化疗方案治疗失败的转移性黑色素瘤患者,接受递增剂量的沙利度胺联合固定剂量塞玛沙尼的治疗。结果:共纳入12例患者,接受了44个疗程的塞马沙尼(Semaxanib)治疗,固定剂量为145 mg/m²,每周两次静脉注射,联合沙利度胺治疗。沙利度胺起始剂量为每日200 mg,根据患者耐受情况逐渐增加剂量。在第一个疗程中,塞马沙尼的给药时间比沙利度胺早1天,以便评估塞马沙尼单独用药(第1疗程)和与沙利度胺联合用药(第2疗程)的药代动力学特征。主要毒性反应包括深静脉血栓形成、头痛和下肢水肿。在10例可评估疗效的患者中,观察到1例完全缓解,持续20个月;1例部分缓解,持续12个月。此外,4例患者病情稳定,持续2至10个月。塞马沙尼的药代动力学特征表现为药物暴露参数与单药II期研究中观察到的参数相当,表明不存在显著的药物相互作用。塞马沙尼血浆峰浓度在第1疗程为1.2-3.8 μg/ml,在第2疗程为1.1-3.9 μg/ml。平均末端半衰期为1.3 (± 0.31) 小时。生物学研究显示,在接受研究超过4个月的患者中,治疗后血清VEGF浓度升高。 结论:塞马沙尼联合沙利度胺治疗方案可行,并显示出对既往治疗失败的转移性黑色素瘤患者的抗肿瘤活性。对于晚期黑色素瘤和其他恶性肿瘤患者,可能需要进一步评估靶向多种血管生成通路的治疗策略。 [5] 背景:这项 I 期研究评估了 Semaxanib/SU5416(一种强效且选择性的血管内皮生长因子 (VEGF) 受体酪氨酸激酶 Flk-1 抑制剂)联合每周一次顺铂和伊立替康治疗晚期实体瘤患者的安全性。 方法:患者从第 1 周至第 4 周每周接受顺铂 30 mg/m² 和伊立替康 50 mg/m² 治疗,同时每周两次接受 SU5416 治疗,剂量分别为 65 mg/m²(剂量水平 (DL)1)或 85 mg/m²(DL2),持续 6 周(1 个疗程)。进行系列 ¹⁸氟代脱氧葡萄糖正电子发射断层扫描 (¹⁸FDG-PET) 和 ¹⁵O-H₂O-PET 扫描。结果:共13例患者接受治疗(7例接受DL1剂量,6例接受DL2剂量);7例患者完成了至少一个疗程的治疗。DL2剂量组3例患者出现剂量限制性毒性(DLT)(3级中性粒细胞减少症和3级血小板减少症导致治疗延迟,3级恶心/呕吐)。DL1剂量组未观察到客观缓解,该剂量被确定为最大耐受剂量(MTD)。DL2剂量组观察到1例部分缓解(PR)。记录了¹⁸FDG-PET显像结果,但根据实体瘤疗效评价标准(RECIST),该显像结果无法预测疗效。结论:SU5416 65 mg/m²,每周两次,联合顺铂和伊立替康,每周一次,共6周,其中4周为一个疗程,耐受性良好,但未观察到临床疗效。 ¹⁸FDG-PET 可能是一种有用的 SU5416 生物活性药效学标志物,但仍需进一步开发。[6] 塞玛沙尼 (SU-5416) 是一种小分子抑制剂,其抗肿瘤作用主要通过阻断 VEGF 介导的血管生成来实现,因为它对肿瘤细胞的直接细胞毒性极低。[1] 塞玛沙尼 (SU-5416) 在动物模型中诱发肺动脉高压的能力凸显了其潜在的心血管毒性,因此在临床应用中需要密切监测。[3] 临床研究表明,塞玛沙尼 (SU-5416) 单药治疗晚期实体瘤的疗效有限,但与化疗或其他靶向药物联合使用时可能具有协同作用。[5,6] |
| 分子式 |
C15H14N2O
|
|---|---|
| 分子量 |
238.28
|
| 精确质量 |
238.11
|
| 元素分析 |
C, 75.61; H, 5.92; N, 11.76; O, 6.71
|
| CAS号 |
204005-46-9
|
| 相关CAS号 |
(Z)-Semaxanib;194413-58-6; 204005-46-9; 1055412-47-9 (Semaxanib analog/chlorinated)
|
| PubChem CID |
5329098
|
| 外观&性状 |
Yellow to orange solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
481.4±45.0 °C at 760 mmHg
|
| 闪点 |
244.9±28.7 °C
|
| 蒸汽压 |
0.0±1.2 mmHg at 25°C
|
| 折射率 |
1.684
|
| LogP |
2.87
|
| tPSA |
44.89
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
1
|
| 可旋转键数目(RBC) |
1
|
| 重原子数目 |
18
|
| 分子复杂度/Complexity |
377
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O=C1/C(=C(/[H])\C2=C(C([H])([H])[H])C([H])=C(C([H])([H])[H])N2[H])/C2=C([H])C([H])=C([H])C([H])=C2N1[H]
|
| InChi Key |
WUWDLXZGHZSWQZ-WQLSENKSSA-N
|
| InChi Code |
InChI=1S/C15H14N2O/c1-9-7-10(2)16-14(9)8-12-11-5-3-4-6-13(11)17-15(12)18/h3-8,16H,1-2H3,(H,17,18)/b12-8-
|
| 化学名 |
(3Z)-3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylidene]-1H-indol-2-one
|
| 别名 |
Sugen 5416; Sugen5416; Sugen-5416; semoxind; SU5416; SU-5416; SU 5416; Semaxanib; Semaxinib; 204005-46-9; 194413-58-6
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (10.49 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浮液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: 2.25 mg/mL (9.44 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 22.5 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 View More
配方 3 中的溶解度: 1% DMSO+30% polyethylene glycol+1% Tween 80: 30 mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.1967 mL | 20.9837 mL | 41.9674 mL | |
| 5 mM | 0.8393 mL | 4.1967 mL | 8.3935 mL | |
| 10 mM | 0.4197 mL | 2.0984 mL | 4.1967 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT00004868
Conditions:Brain and Central Nervous System TumorsLink: https://clinicaltrials.gov/ct2/show/NCT00005042
Conditions:SarcomaLink: https://clinicaltrials.gov/ct2/show/NCT00021281
Conditions:Colorectal Cancer
Title:SU5416 Combined With Gemcitabine and Cisplatin in Treating Patients With Advanced Solid Tumors
Status:Unknown status
updateDate:2013-12-19
Ctid:NCT00005996
Link: https://clinicaltrials.gov/ct2/show/NCT00005996
Conditions:Unspecified Adult Solid Tumor, Protocol SpecificLink: https://clinicaltrials.gov/ct2/show/NCT00026377
Conditions:Prostate CancerLink: https://clinicaltrials.gov/ct2/show/NCT00006002
Conditions:Prostate CancerLink: https://clinicaltrials.gov/ct2/show/NCT00006013
Conditions:Multiple Myeloma and Plasma Cell NeoplasmLink: https://clinicaltrials.gov/ct2/show/NCT00026260
Conditions:Cervical CancerLink: https://clinicaltrials.gov/ct2/show/NCT00023725
Conditions:SarcomaLink: https://clinicaltrials.gov/ct2/show/NCT00023738
Conditions:SarcomaLink: https://clinicaltrials.gov/ct2/show/NCT00006001
Conditions:Colorectal CancerLink: https://clinicaltrials.gov/ct2/show/NCT00006014
Conditions:Malignant MesotheliomaLink: https://clinicaltrials.gov/ct2/show/NCT00005862
Conditions:Gastrointestinal Stromal Tumor|SarcomaLink: https://clinicaltrials.gov/ct2/show/NCT00017316
Conditions:Melanoma (Skin)Link: https://clinicaltrials.gov/ct2/show/NCT00006000
Conditions:Unspecified Adult Solid Tumor, Protocol SpecificLink: https://clinicaltrials.gov/ct2/show/NCT00006003
Conditions:Recurrent Melanoma|Stage IV MelanomaLink: https://clinicaltrials.gov/ct2/show/NCT00005818
Conditions:Adenocarcinoma of the Colon|Adenocarcinoma of the Rectum|Recurrent Colon Cancer|Recurrent Rectal Cancer|Stage III Colon Cancer|Stage III Rectal Cancer|Stage IV Colon Cancer|Stage IV Rectal CancerLink: https://clinicaltrials.gov/ct2/show/NCT00005942
Conditions:Chronic Myelomonocytic Leukemia|Previously Treated Myelodysplastic Syndromes|Recurrent Adult Acute Myeloid Leukemia|Refractory Anemia With Excess Blasts|Refractory Anemia With Excess Blasts in TransformationLink: https://clinicaltrials.gov/ct2/show/NCT00009919
Conditions:Recurrent Renal Cell Cancer|Stage IV Renal Cell CancerLink: https://clinicaltrials.gov/ct2/show/NCT00004252
Conditions:Colorectal CancerLink: https://clinicaltrials.gov/ct2/show/NCT00003720
Conditions:SarcomaLink: https://clinicaltrials.gov/ct2/show/NCT00006384
Conditions:Kidney CancerLink: https://clinicaltrials.gov/ct2/show/NCT00005822
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT00005642
Conditions:Unspecified Adult Solid Tumor, Protocol SpecificLink: https://clinicaltrials.gov/ct2/show/NCT00005647
Conditions:Head and Neck CancerLink: https://clinicaltrials.gov/ct2/show/NCT00006257
Conditions:Unspecified Adult Solid Tumor, Protocol SpecificLink: https://clinicaltrials.gov/ct2/show/NCT00006361
Conditions:Carcinoma of Unknown Primary|Head and Neck Cancer|Non-melanomatous Skin CancerLink: https://clinicaltrials.gov/ct2/show/NCT00006247
Conditions:Brain and Central Nervous System TumorsLink: https://clinicaltrials.gov/ct2/show/NCT00002226
Conditions:Sarcoma, Kaposi|HIV InfectionsLink: https://clinicaltrials.gov/ct2/show/NCT00005931
Conditions:Sarcoma, Kaposi|HIV Infections
|
|
|
|
 VEGFR2-IN-7
VEGFR2-IN-7
 SYHA1813
SYHA1813
 VEGFR-2-IN-38
VEGFR-2-IN-38
 BHEP
BHEP
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA




 463611831
463611831
