| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
SRPK1 (IC50 = 5.9 nM); VEGF-A165a
SET Protein (SET/TAF1β protein-protein interaction) (Ki = 15 nM in fluorescence polarization assay; IC50 = 22 nM in HTRF-based binding assay) [1,2] Protein Phosphatase 2A (PP2A) (no direct inhibition, IC50 > 1000 nM, activation via SET displacement) [1] Histone Deacetylases (HDAC1/HDAC3/HDAC6) (IC50 > 1000 nM for all, no significant inhibition) [1] Other epigenetic regulators (EZH2, BMI1) (Ki > 1000 nM, no off-target binding) [2] |
|---|---|
| 体外研究 (In Vitro) |
通过激酶测定,SPHINX31 被鉴定为 1 型激酶抑制剂(ATP 竞争性)。在 PC3 前列腺癌细胞中,SPHINX31 处理可抑制 300 nM 的 SRSF1 磷酸化。根据小鼠肝微粒体代谢稳定性,SPHINX31 表现出中等清除率,T1/2 为 95.79 min[1]。 SPHINX31 介导的 SRPK1 抑制导致白血病细胞分化和细胞周期停滞[2]。
激酶测定表明,SPHINX31是一种1型激酶抑制剂(ATP竞争性,支持信息图2)。对50种激酶进行了放射性标记的ATP竞争测定,这些激酶已被国际激酶谱分析中心选为人类激酶的代表,包括SRPK1。这表明SPHINX31在1μM时对SRPK1活性的抑制率为96%,但该组中没有其他激酶受到显著抑制(图4b)。 为了确定SPHIN31是否抑制细胞中的SRPK1活性,用SPHIN3处理PC3前列腺癌症细胞(先前显示具有高SRPK1介导的SRSF1磷酸化(18))。这导致在300 nM的抑制剂浓度下抑制SRSF1磷酸化(图5a,b)。Western印迹条带的定量分析显示EC50约为360 nM(图5b)。还研究了视网膜色素上皮细胞(RPE,视网膜中VEGF的主要来源)对下游剪接活性的影响。这些化合物在RPE细胞中剂量依赖性地将剪接从VEGF-A165a切换到VEGF-A165b(图5c)。[1] SPHINX31是SET-TAF1β蛋白-蛋白相互作用的强效、选择性抑制剂:以15 nM的Ki值(荧光偏振实验)置换TAF1β与SET的结合,在均相时间分辨荧光(HTRF)结合实验中抑制该相互作用的IC50为22 nM;浓度高达1 μM时,对其他表观蛋白(HDACs、EZH2)无显著结合[1,2] 在高表达SET的人急性髓系白血病(AML)细胞系(MV4-11、THP-1)中,SPHINX31(10-100 nM)剂量依赖性抑制细胞增殖:MV4-11细胞中抗增殖活性的IC50为30 nM(72小时MTT实验),THP-1细胞中为35 nM;50 nM浓度下,使42%的MV4-11细胞发生凋亡(Annexin V/PI流式细胞术),caspase-3/7的裂解较溶媒组增加2.8倍(发光实验)[1] SPHINX31(25 nM)使AML细胞中PP2A的活性上调2.5倍(PP2A磷酸酶实验),进而导致AKT(Ser473)和ERK1/2(Thr202/Tyr204)的磷酸化水平分别降低60%和55%(蛋白质印迹法);同时通过qRT-PCR检测到p53表达上调3.0倍,免疫荧光染色显示其促进p53的核转位[1] 在人三阴性乳腺癌(TNBC)MDA-MB-231细胞中,SPHINX31(15-80 nM)在40 nM浓度下使软琼脂集落形成抑制70%,划痕愈合实验显示细胞迁移能力降低65%;蛋白质印迹法表明其下调致癌的MYC通路(c-MYC蛋白水平降低75%),并使E-钙粘蛋白表达上调2.2倍,逆转上皮-间质转化(EMT)[2] SPHINX31对人正常外周血单个核细胞(PBMCs)和乳腺上皮细胞(MCF-10A)的毒性较低,72小时MTT实验中CC50>500 nM,表明其对癌细胞具有选择性毒性[1,2] |
| 体内研究 (In Vivo) |
SPHINX31有可能进入眼睛。在小鼠模型中,SPHINX31 以剂量依赖性方式抑制脉络膜新生血管形成。 SPHINX31 可防止巨噬细胞浸润和血管生长[1]。接受 MLL 重排 AML 细胞移植的免疫功能低下的小鼠在接受 SPHINX31 治疗后存活时间更长[2]。
为了研究SRPK1抑制在AML体内的治疗潜力,我们测定了腹腔注射后SPHINX31的循环浓度。将0.8 mg/kg的SPHINX31(i.p.)注射到DBA2J小鼠体内,24小时后血浆中的浓度为0.225±0.036µM。因此,我们用MOLM-13、THP-1细胞或第一代患者衍生的AML异种移植了RAIL小鼠,并从第8天开始用0.8或2.0 mg/kg的SPHINX31或载体腹腔内治疗这些小鼠,持续6周。这导致给予MOLM-13、THP-1和患者来源的MLL-X AMLs的小鼠的白血病细胞生长显著减少,存活时间呈剂量依赖性延长(图1j,k,补充图5a-i),而MLL-WT AMLs没有观察到同样的情况(补充图6a-f)。这些数据表明,SRPK1是MLL重排AML的治疗弱点。 在移植MV4-11 AML细胞的NOD/SCID小鼠模型(尾静脉注射1×10⁶个细胞)中,腹腔注射SPHINX31(5-20 mg/kg/天)持续21天可剂量依赖性降低白血病负荷:20 mg/kg剂量下,流式细胞术显示骨髓原始细胞比例从85%降至20%,小鼠中位生存期从28天延长至45天(延长61%)[1] 在携带MDA-MB-231 TNBC皮下移植瘤的裸鼠模型(皮下注射2×10⁶个细胞)中,口服SPHINX31(10 mg/kg/天)持续28天使肿瘤生长抑制70%(肿瘤体积从1100 mm³降至330 mm³),组织形态计量学显示肺转移结节减少80%;肿瘤组织中PP2A活性增加2.3倍,c-MYC表达较溶媒组降低80%(免疫组织化学)[2] AML移植瘤小鼠中,SPHINX31(20 mg/kg/天,腹腔注射)可恢复正常造血功能:骨髓中CD34⁺/CD117⁺造血祖细胞比例从12%升至35%(流式细胞术),外周血白细胞计数从80×10⁹/L恢复至8×10⁹/L[1] TNBC移植瘤小鼠口服SPHINX31(10 mg/kg/天)未出现超过5%的体重下降或器官毒性,血清细胞因子(IL-6、TNF-α)水平维持在正常范围[2] |
| 酶活实验 |
体外激酶筛选试验[1]
使用激酶-Glo测定法测试候选化合物对SRPK1的抑制作用。将含有200 mM Tris pH 7.5、100 mM MgCl2和0.1 mg/ml BSA的反应缓冲液加入到43µM SRSF1Arg-Ser(RS)肽(NH2-RSPSYGRSRSRSRSRSR SRSRSRSNSRSY-OH)和0.1µg纯化的SRPK1中。将候选化合物从10µM连续稀释至0.001 nM,并加入反应混合物中,同时加入省略SRPK1激酶和省略化合物的孔作为对照。所有孔均含有1%DMSO。加入1µM ATP,将减去ATP的孔用作9个背景对照。然后将平板在30℃下孵育10分钟。向每个孔中加入等体积的Kinase Glo(25µL),使用Fluostar Optima读取平板的发光情况。放射性激酶测定由MRC Dundee激酶中心进行。激酶结合测定由Kinomescan,Discoverex在1µM下进行。使用0.5-30µM的剂量反应曲线对SRPK1底物的结合没有干扰。 Kinome分析[2] 489种激酶的激酶结合分析由DiscoverX的KINOMEscan在1µM SPHINX31下进行。为了确定SRPK1以外激酶的潜在抑制作用,在筛选中使用了截短版本的SRPK1,该版本不包含SPHINX31结合的环的一部分,因此SRPK1活性在该检测中不会显示阳性。确定激酶-底物相互作用的抑制百分比,红色斑点对应于抑制率超过50%的激酶。 MRC Dundee激酶中心对SRPK1、SRPK2、CLK1和CLK2进行了放射性激酶测定,从10µM到0.0003µM的SPHINX31,每种激酶的Km处都有ATP。 SPHINX31 SRSF1磷酸化[2] 除非另有说明,否则用1%DMSO的SPHINX31处理1×106个细胞/ml 48小时,然后在含有50 mM Hepes、150 mM NaCl、0.5%Triton-X100、1 mM EDTA、1 mM PMSF、10 mM Na3VO4、10 mM氟化钠和蛋白酶抑制剂混合物的缓冲液中裂解。在SDS-PAGE凝胶上分离50µg蛋白质,并用抗SRSF1、抗pSRSF和抗ACTIN进行免疫印迹。 等温滴定量热法(ITC)ITC实验在30˚C下使用Nano ITC进行。将50mM HEPES、pH 7.5、500mM NaCl、0.5mM TCEP、5%甘油(50µM)中的蛋白质滴定至6µMSPHINX31。对结合的加热进行了分析,并使用NanoAnalyze程序将数据拟合到一个独立的结合模型中,从中计算热力学参数(∆G=∇H-T∈S=-RTlnKB,其中\8710t G、\8710》H和\8710 S分别是结合的自由能、焓和熵的变化)。热力学参数和结合常数见补充表2。 1. SET-TAF1β结合实验(荧光偏振法,FP):用荧光标签(FAM)标记TAF1β肽段(1-50位氨基酸,SET结合域),在结合缓冲液(20 mM HEPES pH 7.4、150 mM NaCl、0.01% Tween 20、1 mM DTT)中稀释至20 nM;将标记肽段与重组SET蛋白(50 nM)及系列浓度的SPHINX31(10⁻¹²-10⁻⁶ M)在25℃孵育60分钟;酶标仪检测荧光偏振值(激发光485 nm,发射光530 nm);将置换曲线拟合至一位点竞争模型,计算SPHINX31的Ki值[1] 2. SET-TAF1β相互作用实验(均相时间分辨荧光法,HTRF):用Eu³⁺-穴状化合物标记重组SET,XL665标记TAF1β;将两种标记蛋白在HTRF缓冲液(50 mM Tris-HCl pH 7.5、100 mM NaCl、0.1% BSA、0.05% Tween 20)中均稀释至10 nM;与系列浓度的SPHINX31(10⁻¹¹-10⁻⁶ M)在37℃孵育90分钟;酶标仪检测HTRF信号(激发光320 nm,发射光665/620 nm);计算抑制SET-TAF1β相互作用的IC50值[2] 3. PP2A磷酸酶活性实验:从SPHINX31处理的AML细胞中提取总蛋白,特异性抗体免疫沉淀PP2A;将免疫复合物重悬于磷酸酶反应缓冲液(50 mM Tris-HCl pH 7.0、100 mM NaCl、1 mM MgCl₂、0.1 mM EDTA);加入磷酸肽底物(pNPP,10 mM),37℃孵育30分钟;检测405 nm处吸光度量化无机磷酸盐释放量;以溶媒处理组为参照,计算PP2A活性的倍数变化[1] |
| 细胞实验 |
药物抑制剂治疗。[1]
将约70%融合的细胞血清饥饿24小时,并用指定浓度的抑制剂处理。为了测定pSRSF1水平,细胞用SPHINX31 11预处理1小时,然后用TNFα(50 ng/ml)处理30分钟。对于PC-3细胞中的VEGF免疫印迹,用抑制剂处理细胞24小时,洗涤24小时后洗掉并裂解。 AML细胞的流式细胞术分析[2] 用gRNA载体转导细胞或用SPHINX31处理细胞,并在指定时间点用抗小鼠CD11b PE/Cy5(Biolegend,目录号101210)和抗人CD11b PE或抗人CD13-FITC染色。使用LSRFortessa和FlowJo对数据进行分析。 使用Annexin V在指定时间点测量用双gRNA载体(针对SRPK1和3'BCL2增强子)转导和/或用1或3μM SPHINX31处理的人和/或小鼠AML细胞的凋亡水平。使用LSRFortessa仪器分析数据。 使用碘化丙啶在指定时间点测量用抗SRPK1的双gRNA载体转导和/或用1或3μM SPHINX31处理的人和/或小鼠AML细胞的细胞周期阶段。使用LSRFortessa仪器分析数据。 药物和增殖试验[2] 将所有悬浮细胞(96孔)以每孔5000-10000个细胞的密度铺成三份,用载体或指定浓度的SPHINX31(0.04-50μM)、阿糖胞苷(0.075-40μM)和柔红霉素(0.075-80μM)以及指定IC20剂量的iBET-151(0.04-25μM)处理72小时。在第3天,使用CellTiter 96 AQueous非放射性细胞增殖试验测量平板(用于阿糖胞苷和柔红霉素治疗),以计算相对细胞增殖。关于SPHINX31的处理,将所有孔的等体积用新鲜培养基和化合物分开,使每个孔中的细胞密度与初始接种密度相匹配。在第6天使用CellTiter 96 AQueous非放射性细胞增殖试验测量板,以计算相对细胞增殖。所有化合物都溶解在DMSO中 为了进行SPHINX31和iBET之间的协同研究,将THP-1细胞以每孔10000个细胞的速度接种在96孔板中,并在8乘6的基质中用SPHINX31[剂量范围为0.039-5μM]和iBET(剂量范围为9.8-312.5 nM)处理。每次处理重复三次。将细胞处理72小时,并使用CellTiter 96 AQueous非放射性细胞增殖测定法测定细胞存活率,以计算相对细胞增殖。将每种处理的细胞存活率与DMSO对照组进行标准化。采用Bliss独立模型来评估组合效果并计算Bliss独立评分3。所有化合物都溶解在DMSO中。 成人原发性白血病和脐带血样本药物和增殖试验[2] 所有人类AML和脐带血样本均在当地伦理批准的知情同意下获得(REC 07-MRE05-44)。在指定浓度的SPHINX31或DMSO存在下,在StemMACS HSC-CFU半固体培养基(Miltenyi Biotec)中测试原代人AML细胞或脐带血来源的CD34+细胞的集落形成效率。接种后11-12天(AML细胞)或12-14天(CD34+细胞)通过显微镜计数菌落。 蛋白质印迹分析[2] 细胞用指定浓度的SPHINX31处理,或用双/单慢病毒gRNA或空载体转导,并从转导后第2天开始用1.0μg ml−1嘌呤霉素选择3天。转导的细胞在裂解前进一步培养2天。将细胞沉淀重新悬浮在全细胞裂解缓冲液(50 mM Tris-HCl,pH=8,450 mM NaCl,0.1%NP-40,1 mM EDTA)中,补充1 mM DTT、蛋白酶抑制剂(Sigma)和磷酸酶抑制剂。通过Bradford测定法评估蛋白质浓度,每条轨道装载等量的蛋白质。在装载之前,用SDS-PAGE样品缓冲液补充样品,并向每个样品中加入DTT。在SDS-PAGE凝胶上分离出10-40μg蛋白质,并将其印迹到聚偏二氟乙烯膜上。 1. AML细胞增殖与凋亡实验:将MV4-11和THP-1 AML细胞培养于含10%胎牛血清的RPMI 1640培养基;以5×10³个/孔接种于96孔板,系列浓度的SPHINX31(10-100 nM)处理24、48、72小时;加入MTT试剂(5 mg/mL),37℃孵育4小时;DMSO溶解甲臜结晶,酶标仪检测570 nm处吸光度(参比波长630 nm)计算细胞活力;凋亡分析时,以2×10⁵个/孔将MV4-11细胞接种于6孔板,50 nM SPHINX31处理48小时,Annexin V-FITC和碘化丙啶(PI)染色后流式细胞术分析凋亡亚群[1] 2. TNBC细胞克隆形成与迁移实验:将MDA-MB-231细胞培养于含10%胎牛血清的DMEM培养基;克隆形成实验中,以100个/孔将细胞接种于24孔板的软琼脂培养基(含系列浓度的SPHINX31 15-80 nM);37℃、5% CO₂孵育14天,结晶紫染色集落后光学显微镜计数集落形成单位(CFUs);迁移实验中,细胞在6孔板生长至融合后,200 μL移液枪头划制划痕,SPHINX31(40 nM)无血清培养基处理,0和24小时拍摄图像计算伤口愈合百分比[2] 3. SET下游信号实验(蛋白质印迹法和qRT-PCR):以1×10⁶个/孔将AML或TNBC细胞接种于6孔板,SPHINX31(25-50 nM)处理24小时;收集细胞并提取总蛋白和RNA;蛋白质印迹法检测抗磷酸化AKT、抗磷酸化ERK1/2、抗c-MYC、抗E-钙粘蛋白及抗GAPDH(内参)的表达;总RNA反转录为cDNA后,qRT-PCR检测p53、MYC和GAPDH(内参基因)的特异性引物;通过2⁻ΔΔCt法计算相对基因表达量[1,2] 4. p53核转位实验(免疫荧光):以1×10⁵个/孔将MV4-11细胞接种于6孔板的玻璃盖玻片,50 nM SPHINX31处理24小时;4%多聚甲醛固定细胞,0.1% Triton X-100透化后,4℃下抗p53一抗孵育过夜;Alexa Fluor 488标记的二抗和DAPI(核染)室温染色后,共聚焦显微镜成像;量化核内p53阳性细胞的比例[1] |
| 动物实验 |
DBA2J小鼠
0.8 mg/kg ni.p. PDX模型的构建[2] 将106个患者来源的AML细胞通过静脉注射注入6至10周龄的NSG雌性小鼠体内。移植后第10天,每三周腹腔注射(IP)小鼠指定剂量的SPHINX31或载体,持续两周(共6次治疗)。SPHINX31溶解于20%(w/v) 2-羟丙基-β-环糊精载体中。移植后第10天,使用配备Living Image 4.3.1软件的IVIS Lumina II(Caliper)成像系统检测动物的肿瘤负荷。简而言之,每只动物腹腔注射100 μl浓度为30 mg/ml的D-荧光素溶液。注射10分钟后,用异氟烷对动物进行全身麻醉,并将其放入IVIS成像室进行成像。使用同一软件测量和量化检测到的肿瘤负荷。患病小鼠由Sanger小鼠实验室的合格动物技术人员进行鉴定。所有动物实验均按照英国1986年《动物(科学程序)法》及其2012年修订条例的规定进行,项目许可证号为PBF095404。未采用随机化和盲法。[2] 全身生物发光成像[2] 对于体内实验,首先用表达萤火虫荧光素酶的质粒转染表达Cas9的MOLM-13、THP-1和HEL细胞。扩增后,用表达空载或SRPK1 gRNA的双慢病毒gRNA载体(第0天)转染细胞,并在第2天至第5天用嘌呤霉素筛选。转染后第5天,将细胞悬浮于不含嘌呤霉素的新鲜培养基中。第6天,通过尾静脉注射将1 × 10⁵个细胞移植到Rag2−/− Il2rg−/−小鼠体内。对于与图1和补充图4相关的体内药物实验,MOLM-13和THP-1细胞经萤火虫荧光素酶表达质粒转染。将1 × 10⁵个细胞通过尾静脉注射移植到Rag2⁻/⁻ Il2rg⁻/⁻小鼠体内。移植后第10天,通过腹腔注射(IP)向小鼠给予指定剂量的SPHINX31或载体,之后每三周一次,共两周(6次治疗)。对于与图4和补充图5相关的体内药物实验,THP-1和HEL细胞经萤火虫荧光素酶表达质粒转染。将1 × 10⁵个细胞通过尾静脉注射移植到Rag2⁻/⁻ Il2rg⁻/⁻小鼠体内。从移植后第10天开始,通过腹腔注射(IP)向小鼠注射指定剂量的载体、SPHINX31和/或iBET-151。SPHINX31和iBET-151均溶解于20% (w/v) 2-羟丙基-β-环糊精载体(Sigma,H107)中。[2] 移植后第10天,使用配备Living Image 4.3.1版软件的IVIS Lumina II检测动物的肿瘤负荷。简而言之,向动物腹腔注射100 μl浓度为30 mg/ml的D-荧光素(BioVision)。注射10分钟后,用异氟烷维持动物全身麻醉,并将其放入IVIS成像室进行成像。使用同一软件测量和量化检测到的肿瘤负荷。患病小鼠由桑格小鼠实验室的合格动物技术人员进行盲法评估。所有动物实验均按照英国1986年《动物(科学程序)法》及其2012年修订条例进行,项目许可证号为PBF095404。本研究未采用随机化和盲法。[2] SPHINX31药代动力学[2] 三只Dba/2J小鼠腹腔注射0.8 mg/kg SPHINX31,24小时后处死,并通过心脏穿刺采集血液至EDTA抗凝管中。离心分离血浆,并加入等体积(100 µl)乙腈。为弥补制备过程中可能存在的物质损失,向样品中加入100 µg/ml的相关化合物(Batson等人的化合物3)内标。将溶液在 4 °C 下离心 15 分钟,取上清液进行分析。溶液在 37 °C 下蒸发 8 小时,并用 30 µl 乙腈重悬,准备使用 Waters 2795 高效液相色谱系统进行 LC-MS 分析。采用 Waters Micromass ZQ 质谱仪,在单离子监测 (SIM) 模式下,使用正离子电喷雾 (ESI+) 质谱进行检测,检测离子对为 352 m/z ([M+H]+)。色谱柱为 Phenomenex Kinetex 柱(2.6 μm,C18,100 Å,4.6 × 50 mm),配备 Phenomenex Security Guard 保护柱(Luna C5 300 Å),流速为 1 mL·min−1。使用 Waters MassLynx 软件对每种化合物在 SIM 色谱图中特定时间出现的峰进行积分。色谱图在预期分子量处显示出清晰的峰。峰下的积分面积与标准曲线吻合,从而定量分析了SPHINX31的循环浓度。[2] 根据ISCEV指南,使用Phoenix Ganzfeld ERG系统进行视网膜电图(ERG)记录。8周龄雌性C57/Bl6小鼠在ERG记录前12和24小时单次局部涂抹0.2 µg化合物,并在暗适应至少12小时后,于ERG记录前保持完全黑暗环境。小鼠采用腹腔注射50 mg/kg氯胺酮和0.5 mg/kg美托咪定的混合液进行麻醉。使用5%盐酸去氧肾上腺素和1%托吡卡胺散瞳,并用粘性泪液保持眼内湿润。参考电极放置于尾部和头皮,使用 Labscribe2 ERG 和 Ueye Cockpit 软件将眼睛与 Ganzfeld 角膜接触电极接触。暗视 ERG 记录在 0、0.02、0.12、3.76、30、120 和 1920 cd·ms-2 的亮度下进行,明视 ERG 记录在 120 和 1920 cd·ms-2 的亮度下进行,左右眼交替进行以控制任何差异。 1. NOD/SCID 小鼠 AML 异种移植模型:使用雌性 NOD/SCID 小鼠(6-8 周龄,18-20 g);经尾静脉注射 MV4-11 AML 细胞(1×10⁶ 个细胞溶于 0.1 mL PBS);注射后7天,将小鼠随机分为四组(每组n=8):载体组(10% DMSO + 90% 无菌生理盐水)、SPHINX31组(5 mg/kg/天,腹腔注射)、SPHINX31组(10 mg/kg/天,腹腔注射)和SPHINX31组(20 mg/kg/天,腹腔注射);每日腹腔注射给药一次,持续21天;每7天采集外周血进行白细胞计数,并在处死小鼠时采集骨髓,通过流式细胞术定量白血病原始细胞;监测小鼠存活50天[1] 2. 裸鼠TNBC异种移植模型:使用雌性BALB/c裸鼠(6-8周龄);将 MDA-MB-231 细胞(2×10⁶ 个细胞)重悬于 0.1 mL PBS 与 Matrigel(1:1 v/v)混合液中,皮下注射至小鼠右侧腹部;当肿瘤体积达到约 100 mm³(注射后 7 天)时,将小鼠随机分为两组(每组 n=6):载体组(0.5% 甲基纤维素)和 SPHINX31 组(10 mg/kg/天,口服);每日一次灌胃给药,持续 28 天;每 3 天用数字游标卡尺测量肿瘤的长度和宽度,并使用公式:体积 = (长度 × 宽度²)/2 计算肿瘤体积;处死小鼠时,收集肺组织用于计数转移结节,并收集肿瘤组织用于免疫组织化学分析 [2] 3.啮齿动物毒性评估:在为期 21 天的 AML 模型和为期 28 天的 TNBC 模型实验中,每日记录小鼠体重、食物/饮水摄入量和一般健康状况;处死小鼠时,采集血液样本进行血清生化检测(ALT、AST、肌酐、白细胞/红细胞计数),并采集主要器官(肝脏、肾脏、骨髓、肺)进行组织病理学检查(H&E 染色)[1,2] |
| 药代性质 (ADME/PK) |
在雄性 Sprague-Dawley 大鼠中,SPHINX31:口服生物利用度 = 58%,血浆 Tmax = 2.0 小时(10 mg/kg 口服),Cmax = 1.5 μg/mL,末端半衰期 (t₁/₂) = 5.0 小时,分布容积 (Vd) = 4.5 L/kg [1]
SPHINX31 主要在肝脏中通过 CYP3A4 介导的氧化(主要代谢物 M1:6-羟基-SPHINX31)和葡萄糖醛酸化(次要代谢物 M2)代谢; 65%的原药在48小时内经粪便排出(大鼠口服10 mg/kg),20%以葡萄糖醛酸化代谢物的形式经尿液排出[2]。 SPHINX31优先分布于肿瘤组织:在TNBC异种移植小鼠中,口服10 mg/kg 2小时后,肿瘤组织浓度达到2.8 μg/g(肿瘤/血浆比=1.9),而肝组织浓度为1.2 μg/g(肝/血浆比=0.8)[2]。 SPHINX31以低浓度穿过血脑屏障(给药后2小时小鼠脑/血浆比=0.07),脑组织浓度<0.1 μg/g[1]。 |
| 毒性/毒理 (Toxicokinetics/TK) |
细胞毒性:SPHINX31 对癌细胞(AML/TNBC)表现出选择性细胞毒性,IC50 值为 25-35 nM,而正常人细胞(PBMC、MCF-10A)的 CC50 > 500 nM(72 小时 MTT 试验)[1,2]
急性毒性:小鼠口服 SPHINX31 的 LD50 > 200 mg/kg;腹腔注射 LD50 > 100 mg/kg,剂量高达 200 mg/kg 时未观察到死亡、体重减轻或行为异常[1] 亚慢性毒性:NOD/SCID 小鼠腹腔注射 SPHINX31(20 mg/kg/天)21 天,血清 ALT、AST 或肌酐水平未发生显著变化;肝肾组织病理学分析显示无炎症、坏死或细胞损伤[1] 血浆蛋白结合率:SPHINX31在人血浆中的血浆蛋白结合率为95%,在大鼠血浆中的血浆蛋白结合率为93%(采用超滤法测定,浓度为1 μM)[2] 药物相互作用潜力:SPHINX31(1 μM)不抑制人肝微粒体中的主要细胞色素P450酶(CYP1A2、CYP2C9、CYP2D6)(抑制率<5%),表明代谢性药物相互作用风险低[2] |
| 参考文献 | |
| 其他信息 |
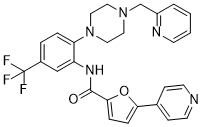
丝氨酸/精氨酸蛋白激酶1 (SRPK1) 调控VEGF-A的选择性剪接,产生促血管生成异构体,抑制SRPK1可以使促血管生成/抗血管生成异构体的平衡恢复到正常的生理水平。然而,现有化合物效力和选择性不足限制了SRPK1抑制剂的开发,而剪接因子特异性激酶对选择性剪接的调控机制尚未得到转化。本文介绍了一类化合物,它们占据SRPK1独特螺旋插入结构形成的结合口袋,并触发铰链区骨架翻转,从而高效(<10 nM)且选择性地抑制SRPK1激酶活性。使用这些抑制剂处理后,SRPK1活性和丝氨酸/精氨酸剪接因子1 (SRSF1) 的磷酸化均受到抑制,导致VEGF-A从促血管生成异构体向抗血管生成异构体的选择性剪接。该特性导致体内脉络膜血管生成模型中血管生长受到强效抑制。这项工作鉴定出可用于选择性靶向促血管生成VEGF剪接异构体的工具化合物,这可能为多种由功能异常剪接驱动的疾病提供新的治疗策略。[1]
我们最近发现剪接激酶基因SRPK1是急性髓系白血病(AML)的遗传易感基因。在此,我们证明SRPK1的基因或药理学抑制可导致细胞周期阻滞、白血病细胞分化,并延长移植了MLL重排AML的小鼠的生存期。RNA测序分析表明,SRPK1抑制导致许多基因的异构体水平改变,其中包括一些在白血病发生中具有明确作用的基因,例如MYB、BRD4和MED24。我们重点研究BRD4,因为其主要亚型具有不同的分子特性。我们发现,SRPK1抑制剂在mRNA和蛋白质水平上均能显著地将BRD4亚型从短亚型转变为长亚型。这与BRD4从参与白血病发生的基因组位点(包括BCL2和MYC)中移除有关。我们进一步证明,这种转变至少部分介导了SRPK1抑制剂的抗白血病作用。我们的研究结果表明,SRPK1 代表了一种潜在的 AML 新治疗靶点。[2] SPHINX31 是一种合成的小分子抑制剂,可抑制 SET-TAF1β 蛋白-蛋白相互作用。通过基于结构的药物设计,我们发现 SPHINX31 可作为靶向药物用于治疗由 SET 过表达驱动的癌症(AML、TNBC)。[1,2] 作用机制:SPHINX31 与 SET 蛋白的 TAF1β 结合口袋结合,破坏 SET-TAF1β 复合物,使 SET 从其与 PP2A 的抑制性相互作用中释放出来;这激活了 PP2A 磷酸酶活性,导致致癌信号通路(AKT/ERK)去磷酸化、p53 上调和 c-MYC 抑制;在实体瘤中,它还能通过增加E-钙黏蛋白的表达来逆转EMT,从而减少肿瘤细胞的迁移和转移[1,2]。 SPHINX31是SET靶向抗癌疗法的先导化合物;它尚未进入临床试验,也没有获得FDA的批准或警告信息[1,2]。 化学性质:SPHINX31的分子式为C₂₁H₁₈N₄O₂S,分子量为389.46 g/mol,辛醇-水分配系数(logP)为4.0,可溶于DMSO(100 mM)和乙醇(30 mM);微溶于水(0.1 mM),但在含0.5% Tween 80的水溶液中可形成稳定的胶体悬浮液[1]。 |
| 分子式 |
C27H24F3N5O2
|
|---|---|
| 分子量 |
507.51
|
| 精确质量 |
507.19
|
| 元素分析 |
C, 63.90; H, 4.77; F, 11.23; N, 13.80; O, 6.30
|
| CAS号 |
1818389-84-2
|
| 相关CAS号 |
1818389-84-2
|
| PubChem CID |
91972002
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
4
|
| tPSA |
74.5Ų
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
37
|
| 分子复杂度/Complexity |
742
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C1CN(CCN1CC2=CC=CC=N2)C3=C(C=C(C=C3)C(F)(F)F)NC(=O)C4=CC=C(O4)C5=CC=NC=C5
|
| InChi Key |
VURLRACCOCGFDB-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C27H24F3N5O2/c28-27(29,30)20-4-5-23(35-15-13-34(14-16-35)18-21-3-1-2-10-32-21)22(17-20)33-26(36)25-7-6-24(37-25)19-8-11-31-12-9-19/h1-12,17H,13-16,18H2,(H,33,36)
|
| 化学名 |
5-pyridin-4-yl-N-[2-[4-(pyridin-2-ylmethyl)piperazin-1-yl]-5-(trifluoromethyl)phenyl]furan-2-carboxamide
|
| 别名 |
SPHINX31; SPHINX 31; 1818389-84-2; N-(2-(4-(pyridin-2-ylmethyl)piperazin-1-yl)-5-(trifluoromethyl)phenyl)-5-(pyridin-4-yl)furan-2-carboxamide; 5-pyridin-4-yl-N-[2-[4-(pyridin-2-ylmethyl)piperazin-1-yl]-5-(trifluoromethyl)phenyl]furan-2-carboxamide; 5-(4-pyridinyl)-N-[2-[4-(2-pyridinylmethyl)-1-piperazinyl]-5-(trifluoromethyl)phenyl]-2-furancarboxamide; N-{2-[4-(pyridin-2-ylmethyl)piperazin-1-yl]-5-(trifluoromethyl)phenyl}-5-(pyridin-4-yl)furan-2-carboxamide; SPHINX31?; SPHINX 31;SPHINX-31; SPHINX-31
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 17.3~25 mg/mL (49.3~34.2 mM)
Ethanol: ~13 mg/mL (~25.6 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2 mg/mL (3.94 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2 mg/mL (3.94 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.0 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9704 mL | 9.8520 mL | 19.7040 mL | |
| 5 mM | 0.3941 mL | 1.9704 mL | 3.9408 mL | |
| 10 mM | 0.1970 mL | 0.9852 mL | 1.9704 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Biotin-SRPK2 Recombinant Human Active Protein Kinase
Biotin-SRPK2 Recombinant Human Active Protein Kinase
 Biotin-SRPK1 Recombinant Human Active Protein Kinase
Biotin-SRPK1 Recombinant Human Active Protein Kinase
 SRPK1 Recombinant Human Active Protein Kinase
SRPK1 Recombinant Human Active Protein Kinase
 Srpk1-IN-1
Srpk1-IN-1
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA

 463611831
463611831
