| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

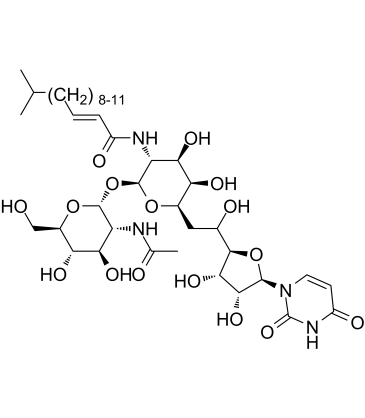
描述:衣霉素是一种同源核苷混合物,是一种含有N-乙酰氨基葡萄糖的抗生素,存在于溶菌链霉菌(Streptomyces lysosuperijkus)中,可抑制蛋白质糖基化。衣霉素会导致未折叠蛋白在细胞内质网(ER)中积累,诱导内质网应激,并导致DNA合成阻滞和细胞周期停滞于G1期。衣霉素可抑制革兰氏阳性菌、酵母、真菌和病毒,并具有抗癌活性。
| 靶点 |
homologous nucleoside antibiotic; N-linked glycosylation; GlcNAc phosphotransferase (GPT)
DPAGT1 (UDP-GlcNAc-dolichol-phosphate N-acetylglucosamine-1 phosphate transferase) [2] GRP78 (glucose-regulated protein-78) – upregulation of GRP78 expression contributes to resistance [1] Skp2 – downregulation of Skp2 leads to p27 accumulation [3] |
|---|---|
| 体外研究 (In Vitro) |
在CD44+/CD24-和原代MCF7细胞中,衣霉素(2 μg/mL;24小时)处理可增强片段相关XBP-1、ATF6核转位水平以及CHOP蛋白表达[1]。研究结果表明,CD44+/CD24-和富含CD44+/CD24的MCF7细胞培养物对衣霉素表现出以下作用:降低细胞迁移、增加细胞死亡和阻断细胞迁移[1]。在Hep3B人肝癌细胞中,衣霉素(1 μg/mL)显著抑制拓扑异构酶抑制剂喜树碱(3 μM)和依托泊苷(5 μM)诱导的细胞凋亡,但对微管蛋白靶向药物紫杉醇(0.1 μM)和长春新碱(0.1 μM)诱导的细胞凋亡无影响。衣霉素增加G1期细胞比例(G1期阻滞)并减少亚G1期细胞比例。衣霉素不改变拓扑异构酶(TOP I、TOP IIα、IIβ)水平,也不抑制喜树碱和依托泊苷引起的ATM激活。衣霉素阻断了喜树碱和依托泊苷诱导的Bcl-2家族蛋白(Bid、Bad、Bax)的裂解以及caspase-3和caspase-7的激活。衣霉素还阻断了喜树碱诱导的细胞周期蛋白E、细胞周期蛋白A和Cdk2的上调,以及依托泊苷诱导的细胞周期蛋白A和Cdk2的增加。单独使用衣霉素可诱导时间依赖性的G1期阻滞。后续添加实验表明,在拓扑异构酶抑制剂处理后较晚添加衣霉素可降低其抗性效应。姜黄素(30 μM)是另一种G1期阻滞诱导剂,它同样抑制了喜树碱和依托泊苷诱导的细胞凋亡。利用 siRNA(序列:5'-AUA ACA UUU AGG CCA GCA AUA GUU C-3')敲低 GRP78 可部分逆转衣霉素诱导的喜树碱耐药性,但并不影响衣霉素诱导的细胞周期调节因子抑制,表明 GRP78 和 G1 期阻滞是独立的因素 [1]。
在多种肝细胞癌细胞系(MHCC-97L、MHCC-97H、Huh7、SMMC-7721、PLC/PRF/5、HCC-LY5)中,衣霉素(2.5 μg/ml,处理 24 小时)以时间和剂量依赖的方式显著降低了 ABCG2 蛋白的表达,并抑制了 Akt 的磷酸化。衣霉素(2.5 μg/ml,处理 24 小时)改变了 ABCG2 的亚细胞定位,使其从质膜转移至细胞内聚集。衣霉素降低了与ABCG2外排活性相关的侧群(SP)率。衣霉素(1.0 μg/ml,处理48小时)降低了癌症干细胞(CSC)标志物(CD133、CD44、CD13、EpCAM、CK-19)的表达,并降低了PCNA的表达,同时增加了裂解型PARP的水平。与单药治疗相比,衣霉素(1.0 μg/ml)与5-氟尿嘧啶(5-FU,MHCC-97L细胞中为100 μg/ml;Huh7细胞中为10 μg/ml)或顺铂(MHCC-97L细胞中为20 μg/ml;Huh7细胞中为3.5 μg/ml)联合治疗48小时,可进一步降低ABCG2、p-Akt、PCNA和CSC标志物的表达,并增强裂解型PARP的水平。 ABCG2 过表达或组成型激活的 Akt (Akt-myr) 可部分逆转衣霉素对顺铂的增敏作用 [2]。 在黑色素瘤细胞系(Mel-RM、A375、MEWO、MM200、Mel4405、IGR3)中,衣霉素 (3 μM) 可诱导时间依赖性的 G1 期细胞周期阻滞,并增加 p27 蛋白水平,但不影响细胞周期蛋白 D1 水平。shRNA 敲低 p27 可逆转 G1 期阻滞。衣霉素通过抑制 p27 的泛素依赖性蛋白酶体降解,延长了 p27 的半衰期(从约 4 小时延长至 >8 小时)。衣霉素以时间依赖性方式降低 Skp2 蛋白水平,但不影响 Pirh2。实时 PCR 显示,Skp2 的降低发生在转录水平(mRNA 下调)。使用 GST-p27 与衣霉素处理的细胞裂解物进行体外泛素化分析显示 p27 的多聚泛素化减少 [3]。 |
| 体内研究 (In Vivo) |
在 CD133+/- MHCC97L 细胞异种移植模型(BALB/c (nu/nu) 小鼠)中,tunitoxin(0.1 mg/kg 或 0.5 mg/kg)治疗可显著降低肿瘤生长 [2]。
在 BALB/c (nu/nu) 小鼠异种移植模型中使用 CD133+ MHCC-97L 细胞,用衣霉素(0.5 mg/kg)治疗 4 周后,与对照组相比,肿瘤数量减少(6 只小鼠中有 3 只),肿瘤体积也更小。衣霉素降低了异种移植肿瘤中的 ABCG2 和 p-Akt 蛋白水平 [2]。 在 MHCC-97L 和 Huh7 异种移植裸鼠模型中,与单药治疗相比,衣霉素(正文中未明确指出剂量,但可能是 0.5 mg/kg)与低剂量顺铂联合治疗显著抑制了肿瘤生长,这可以通过肿瘤重量和体积来衡量 [2]。 |
| 酶活实验 |
为了评估ABCG2的N-连接糖基化,根据标准方案,将MHCC-97L和MHCC-97H细胞裂解液在37°C下用PNGase F、Endo Hf或α-2,3-神经氨酸酶处理。通过Western印迹检测ABCG2分子量的变化,证实ABCG2发生了糖基化[2]。
为了进行p27的体外泛素化实验,纯化了在大肠杆菌中表达的GST-p27融合蛋白,并将其固定在谷胱甘肽琼脂糖珠上。然后将结合在珠上的GST-p27与用或不用衣霉素(3 μM,处理30小时,随后用MG132处理6小时)处理的Mel-RM细胞裂解液孵育3小时。孵育后,洗涤磁珠,用抗泛素抗体对洗脱的蛋白质进行蛋白质印迹分析,以检测 p27 的多泛素化形式 [3]。 |
| 细胞实验 |
Western Blot 分析
细胞类型: CD44+/CD24- 和原始 MCF7 细胞[1] 测试浓度: 2 µg/mL 孵育时间: 24 小时 实验结果: 在 CD44+/CD24- 和原始 MCF7 细胞中检测到剪接型 XBP-1 和 ATF6 的核转位,且 CHOP 蛋白表达水平升高。 肝细胞癌对多种抗癌药物具有化疗耐药性。衣霉素是一种 N-糖基化抑制剂,可诱导未折叠蛋白反应,并被广泛用作内质网应激的药理学诱导剂。本研究采用多种设计方法,探讨了肝细胞癌 Hep3B 细胞对喜树碱和依托泊苷的耐药机制。衣霉素显著抑制了喜树碱或依托泊苷诱导的细胞凋亡。衣霉素既不改变拓扑异构酶水平,也不抑制喜树碱和依托泊苷引起的ATM激活。数据表明,衣霉素诱导的耐药性可能源于药物捕获的拓扑异构酶-DNA复合物和DNA双链断裂的下游事件。喜树碱和依托泊苷导致多种细胞周期调节因子的蛋白表达增加,并诱导Bcl-2家族蛋白的裂解。这些细胞内分子事件可被衣霉素消除。衣霉素后添加实验设计表明,G1期检查点阻滞是耐药机制的一部分。本研究中的另一种G1期阻滞诱导剂姜黄素也能够诱导类似的耐药效应。此外,转染GRP78 siRNA的细胞对衣霉素诱导的细胞凋亡具有部分抵抗力,但对细胞周期调节因子的抑制作用不敏感,这表明GRP78和G1期阻滞是衣霉素诱导耐药机制的两个独立因素。总之,数据表明衣霉素通过上调GRP78和诱导细胞周期G1期阻滞来诱导对拓扑异构酶抑制剂的耐药性。这些发现也提示,化疗药物与中药联合用药可能导致耐药性,因为这些药物会诱导癌细胞内质网应激和/或细胞周期阻滞。[1]Hep3B细胞培养于含10%胎牛血清和青霉素/链霉素的RPMI-1640培养基中。用溶剂(0.1% DMSO)或药物(衣霉素 1 μg/ml;喜树碱 3 μM;依托泊苷 5 μM;紫杉醇 0.1 μM;长春新碱 0.1 μM;姜黄素 30 μM)处理细胞指定时间后,收集细胞,用 70% 乙醇在 4°C 下固定 30 分钟,用 PBS 洗涤,在室温下用磷酸盐-柠檬酸缓冲液孵育 30 分钟,然后重悬于碘化丙啶溶液(含 Triton X-100、RNase 和 PI)中。使用 FACScan 流式细胞仪和 CellQuest 软件分析 DNA 含量。对于蛋白质印迹实验,细胞在冰冷的裂解缓冲液(10 mM Tris-HCl pH 7.4、150 mM NaCl、1 mM EGTA、1 mM PMSF、抑蛋白酶、亮抑蛋白酶、1% Triton X-100)中裂解。取40 μg蛋白质进行10%或15% PAGE电泳分离,然后转移至硝酸纤维素膜,用PBS/5%脱脂奶粉封闭,并用一抗(针对GRP78、TOP I、TOP II、Bcl-2、Bad、Bid、Bax、细胞周期蛋白、Cdks、磷酸化ATM、caspase等)进行免疫印迹,随后用HRP标记的二抗和化学发光法进行检测。对于 siRNA 转染,将汇合度为 50% 的细胞用 Lipofectamine 2000 转染 GRP78 siRNA(5'-AUA ACA UUU AGG CCA GCA AUA GUU C-3')或对照 siRNA,转染 6 小时,然后用试剂处理 [1]。 肝细胞癌细胞系(Huh7、PLC/PRF/5、SMMC-7721、MHCC-97L、MHCC-97H、MHCC-LM3、HCC-LY5)培养于含 10% FBS 的 DMEM 培养基中。对于细胞分选,PLC/PRF/5 细胞用 PE 标记的抗人 CD133/1 抗体进行标记,并通过流式细胞术 (FACS) 分选。其他 CD133+ 细胞比例 <1% 的细胞系使用 EasySep PE 选择试剂盒进行磁性分离。对于实时定量PCR,使用TRIzol提取总RNA,进行逆转录,并使用SYBR Premix Ex Taq和特异性引物(例如,ABCG2、GAPDH)进行PCR。对于Western blotting,裂解细胞,并使用针对ABCG2、Akt、p-Akt、PCNA、cleaved PARP、CD133、CD44等的抗体检测蛋白质。对于免疫染色,将培养玻片上的细胞用衣霉素(2.5 μg/ml)或LY294002(10 mmol/L)处理24小时,然后固定,并用BXP-21抗ABCG2抗体和DAPI染色,最后通过共聚焦显微镜观察。对于SP分析,用Hoechst 33342染色细胞,并通过流式细胞术进行分析。对于慢病毒转染,将ABCG2 cDNA或Akt-myr克隆到pWPXL载体中,在HEK 293T细胞中包装病毒,然后转导靶细胞[2]。 黑色素瘤细胞(Mel-RM、A375、MEWO、MM200、Mel4405、IGR3)的培养方法如前所述。细胞周期分析中,收集细胞,用70%乙醇在4℃下固定过夜,洗涤后,在室温下用含碘化丙啶(40 mg/ml)和RNase A(200 mg/ml)的PBS孵育30分钟,然后进行流式细胞术分析。蛋白质印迹分析中,用抗p27、抗cyclin D1、抗GRP78、抗Skp2、抗Pirh2和抗GAPDH抗体检测细胞裂解液。对于半定量RT-PCR,总RNA经逆转录后,使用p27和Skp2的引物进行扩增。Skp2的实时PCR采用SYBR Green染料,以肌动蛋白作为内参。对于泛素化分析,将转染HA-泛素的细胞用衣霉素(3 μM)处理30小时,再用MG132处理6小时,然后裂解细胞并用抗p27抗体进行免疫沉淀;免疫沉淀物用抗泛素和抗p27抗体进行免疫印迹分析。为了测定半衰期,将细胞用DMSO、MG132或衣霉素预处理28小时,然后用环己酰亚胺(CHX)孵育指定时间,并通过Western blotting分析p27的表达水平[3]。 |
| 动物实验 |
6~8周龄的BALB/c (nu/nu)小鼠按照上海市实验动物伦理委员会制定的实验方案,在标准条件下饲养。实验结束后,处死小鼠,切除肿瘤并称重,然后用福尔马林固定。冷冻肿瘤样本通过Western blotting和实时PCR进行分析。详细信息见补充数据。[2]
6~8周龄的BALB/c (nu/nu)小鼠在标准条件下饲养。异种移植实验中,将MHCC-97L或Huh7细胞皮下注射到小鼠体内(详细信息见补充数据)。为了评估衣霉素对CD133+ CSCs的影响,将CD133+和CD133- MHCC-97L细胞植入裸鼠体内。肿瘤建立后,小鼠接受衣霉素(0.5 mg/kg)治疗4周(给药途径未明确,可能为腹腔注射)。实验结束时,处死小鼠,切除肿瘤,称重,并用福尔马林固定。冷冻肿瘤样本采用Western blotting和实时PCR分析ABCG2和p-Akt的表达[2]。 |
| 参考文献 | |
| 其他信息 |
肝细胞癌(HCC)具有高度化疗耐药性,而ATP结合盒转运蛋白G亚家族成员2(ABCG2)被认为在这种耐药性中起着关键作用。本研究旨在开发有效的治疗策略,以降低HCC中ABCG2的表达水平并克服其化疗耐药性。首先,我们证实了HCC癌细胞系中ABCG2蛋白水平与化疗耐药性呈正相关。ABCG2优先在富含CD133的高耐药性HCC癌干细胞(CSCs)中表达。此外,ABCG2在HCC细胞中发生N-糖基化修饰,这种修饰有助于维持其蛋白稳定性。N-糖基化(NLG)抑制剂衣霉素显著降低了多种HCC癌细胞系中ABCG2的表达,改变了其亚细胞定位,并逆转了其药物外排效应。此外,衣霉素降低了多种癌症干细胞(CSC)标志物的表达水平,并抑制了CD133(+) CSC的致瘤性。衣霉素联合顺铂(CDDP)可抑制增殖细胞核抗原(PCNA)的表达并增加PARP的裂解;ABCG2或Akt-myr的过表达可部分逆转这种效应。与单药治疗相比,联合治疗能更有效地抑制异种移植小鼠的肿瘤生长。最后,CDDP治疗联合UDP-GlcNAc-聚萜醇-磷酸葡萄糖胺-1-磷酸转移酶(DPAGT1)敲低可重现CDDP联合衣霉素治疗的效果。总之,我们的结果表明,衣霉素可能通过靶向DPAGT1/Akt/ABCG2通路逆转肝细胞癌的耐药性,并提高联合治疗的疗效[2]。
内质网 (ER) 中未折叠蛋白的积累会触发未折叠蛋白反应 (UPR),这是一种应激信号通路。UPR 协调内质网分子伴侣的诱导、蛋白质合成的减少以及细胞周期 G1 期的生长停滞。然而,UPR 诱导 G1 期细胞周期停滞的分子机制尚不清楚。本文报道,衣霉素 (TM,一种内质网应激诱导剂) 可激活 UPR 反应,导致黑色素瘤细胞中 p27 的积累和 G1 期细胞周期停滞。p27 的积累是由于 TM 处理抑制了其多聚泛素化及其后续降解。与 p27 稳定相关的是 TM 处理后 p27 E3 连接酶 Skp2 水平的降低。更重要的是,敲低 p27 可显著降低 TM 诱导的 G1 期细胞周期停滞。总之,这些数据表明p27是内质网应激诱导生长停滞的关键介质。[3] 衣霉素是一种N-糖基化抑制剂,可诱导内质网应激和未折叠蛋白反应。它上调GRP78(一种内质网分子伴侣)并导致G1期细胞周期阻滞。在肝细胞癌中,衣霉素可通过上调GRP78和G1期阻滞诱导对拓扑异构酶抑制剂(喜树碱、依托泊苷)的耐药性,提示在将诱导内质网应激的中药与化疗药物联合使用时应谨慎[1]。相反,衣霉素可通过下调ABCG2并经由DPAGT1/Akt/ABCG2通路抑制癌干细胞,从而增强顺铂在肝细胞癌中的疗效,这提供了一种潜在的联合治疗策略[2]。在黑色素瘤细胞中,衣霉素诱导的内质网应激通过转录下调 Skp2 导致 p27 积累,从而引起 G1 期阻滞 [3]。 |
| 分子式 |
C₃₉H₆₄N₄O₁₆(N=₁₀)
|
|---|---|
| 分子量 |
844.94 (n=10)
|
| 精确质量 |
844.94 (n=10)
|
| CAS号 |
11089-65-9
|
| 外观&性状 |
Off-white to light yellow solid powder
|
| 密度 |
1.4±0.1 g/cm3
|
| 熔点 |
234 - 235ºC
|
| 折射率 |
1.617
|
| LogP |
2.58
|
| tPSA |
311.82
|
| InChi Key |
ZHSGGJXRNHWHRS-VIDYELAYSA-N
|
| InChi Code |
InChI=1S/C30H46N4O16/c1-11(2)5-4-6-16(38)32-19-23(43)20(40)14(47-29(19)50-28-18(31-12(3)36)22(42)21(41)15(10-35)48-28)9-13(37)26-24(44)25(45)27(49-26)34-8-7-17(39)33-30(34)46/h4,6-8,11,13-15,18-29,35,37,40-45H,5,9-10H2,1-3H3,(H,31,36)(H,32,38)(H,33,39,46)/b6-4+/t13?,14-,15-,18-,19-,20+,21-,22-,23-,24+,25-,26-,27-,28-,29+/m1/s1
|
| 化学名 |
(E)-N-((2S,3R,4R,5R,6R)-2-(((2R,3R,4R,5S,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)-6-(2-((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-2-hydroxyethyl)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)-5-methylhex-2-enamide
|
| 别名 |
NSC 177382; NSC177382; NSC-177382
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~33.33 mg/mL
H2O : < 0.1 mg/mL |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2 mg/mL (Infinity mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2 mg/mL (Infinity mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.0mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2 mg/mL (Infinity mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
