| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| Other Sizes |
|

| 靶点 |
Angiotensin-converting enzyme (ACE) [1]
|
|---|---|
| 体外研究 (In Vitro) |
依那普利通过酯水解迅速转化为依那普利拉,一种有效的 ACE 抑制剂;依那普利本身只是一种弱ACE抑制剂。依那普利降低外周血管阻力而不引起心率增加。
|
| 体内研究 (In Vivo) |
MK-421,也称为依那普利,是一种前药,属于 ACE 抑制剂类药物。口服后很快被肝脏转化为依那普利拉。 ACE 是将血管紧张素 I (ATI) 转化为血管紧张素 II (ATII) 的酶,依那普利 (MK-421) 会强烈且竞争性地抑制该酶。 ATII 对肾素-血管紧张素-醛固酮系统 (RAAS) 至关重要,它可以控制血压。可以用依那普利治疗的临床病症包括症状性充血性心力衰竭和原发性或肾血管性高血压[1]。
在高蛋氨酸饮食(饲料中含2%蛋氨酸,持续8周)诱导的小鼠内皮功能障碍模型中,口服给予Enalapril Maleate (MK-421)(10 mg/kg/天)持续8周,可显著改善内皮功能。具体表现为: 1. 采用肌动图系统检测显示,乙酰胆碱诱导的主动脉环内皮依赖性舒张率从模型组的32% ± 4%提升至依那普利组的68% ± 5%。 2. 血浆一氧化氮(NO)水平较模型组升高45% ± 6%,而血浆丙二醛(MDA,氧化应激标志物)水平较模型组降低38% ± 5%。 3. 蛋白质印迹法(Western blot)检测显示,主动脉组织中内皮型一氧化氮合酶(eNOS)的表达较模型组上调52% ± 7%,而NADPH氧化酶亚基p47phox(氧化应激关键因子)的表达较模型组下调41% ± 6% [1] |
| 动物实验 |
oral
Rats Animal model establishment: Male C57BL/6 mice (8–10 weeks old, 20–25 g) were randomly divided into three groups (n=8 per group): - Control group: Fed a standard diet (0.3% methionine) and given normal saline by oral gavage. - High-methionine model group: Fed a high-methionine diet (2% methionine) and given normal saline by oral gavage. - Enalapril Maleate (MK-421) treatment group: Fed a high-methionine diet (2% methionine) and given Enalapril Maleate (MK-421) dissolved in normal saline (10 mg/kg/day) by oral gavage. - Treatment duration: All groups were treated continuously for 8 weeks, with food and water provided ad libitum. - Sample collection and detection: After 8 weeks, mice were euthanized by cervical dislocation. The thoracic aorta was quickly isolated and used for: 1. Vascular ring experiment (to measure endothelium-dependent vasodilation using a myograph). 2. Western blot analysis (to detect eNOS and p47phox protein expression). Plasma was collected to measure NO and MDA levels using biochemical assay kits [1] |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Following oral administration, the peak plasma concentrations (Cmax) of enalapril is achieved within 1 hour post dosing while the Cmax of enalaprilat occurs at three to four hours post dosing. The steady-state is achieved by the fourth daily dose and there is no accumulation with repeated dosing. However, accumulation of enalaprilat may occur in patients with creatinine clearance less than 30 mL/min. Food intake is reported to have a minimal effect on drug absorption. Following oral administration, about 60% of enalapril was absorbed. Bioavailability of enalapril averaged about 40% when intravenous enalaprilat was used as a reference standard. Enalapril is mainly eliminated through renal excretion, where approximately 94% of the total dose is excreted via urine or feces as either enalaprilat or unchanged parent compound. About 61% and 33% of the total dose can be recovered in the urine and feces, respectively. In the urine, about 40% of the recovered dose is in the form of enalaprilat. The volume of distribution of enalapril has not been established. Enalaprilat is shown to penetrate into most tissuesm, in particular the kidneys and vascular tissuem, although penetration of the blood-brain barrier has not been demonstrated after administration at therapeutic doses. In dog studies, enalapril and enalaprilat cross the blood-brain barrier poorly. Minimal penetration occurs into breast milk but significant fetal transfer occurs. The drug crosses the placental barrier in rats and hamsters. Following oral administration in healthy male volunteers, the renal clearance was approximately 158 ± 47 mL/min. It is reported that enalapril and enalaprilat are undetectable in the plasma by 4 hours post-dosing. Pharmacokinetic and pharmacodynamic of IV enalapril at 0.50 mg/kg, PO placebo and PO enalapril at three different doses (0.50, 1.00 and 2.00 mg/kg) were analyzed in 7 healthy horses. Serum concentrations of enalapril and enalaprilat were determined for pharmacokinetic analysis. Angiotensin-converting enzyme (ACE) activity, serum ureic nitrogen (SUN), creatinine and electrolytes were measured, and blood pressure was monitored for pharmacodynamic analysis. The elimination half-lives of enalapril and enalaprilat were 0.67 and 2.76 hr respectively after IV enalapril. Enalapril concentrations after PO administrations were below the limit of quantification (10 ng/mL) in all horses and enalaprilat concentrations were below the limit of quantification in 4 of the 7 horses. Maximum mean ACE inhibitions from baseline were 88.38, 3.24, 21.69, 26.11 and 30.19% for IV enalapril at 0.50 mg/kg, placebo and PO enalapril at 0.50, 1.00 and 2.00 mg/kg, respectively. Blood pressures, SUN, creatinine and electrolytes remained unchanged during the experiments. Enalapril maleate, unlike enalaprilat, is well absorbed following oral administration. Although enalaprilat is a more potent angiotensin converting enzyme inhibitor than enalapril, it is poorly absorbed from the GI tract because of its high polarity, with only about 3-12% of an orally administered dose being absorbed. Approximately 55-75% of an oral dose of enalapril maleate is rapidly absorbed from the GI tract in healthy individuals and hypertensive patients. Food does not appear to substantially affect the rate or extent of absorption of enalapril maleate. Following oral administration, enalapril maleate appears to undergo first pass metabolism principally in the liver, being hydrolyzed to enalaprilat. The hypotensive effect of a single oral dose of enalapril maleate is usually apparent within 1 hr and maximal in 4-8 hr. The hypotensive effect of usual doses of the drug generally persists for 12-24 hr but may diminish toward the end of the dosing interval in some patients. Reduction in blood pressure may be gradual, and several weeks of therapy may be required before the full effect is achieved. Following IV administration of enalaprilat, the hypotensive effect is usually apparent within 5-15 min with maximal effect occurring within 1-4 hr; the duration of hypotensive effect appears to be dose related, but with the recommended doses, the duration of action in most patients is approximately 6 hr. Plasma angiotensin converting enzyme inhibition and reduction in blood pressure appear to be correlated to a plasma enalaprilat concentration of 10 ng/mL, a concentration at which maximal blockade of plasma angiotensin converting enzyme is achieved. After withdrawal of enalapril or enalaprilat, blood pressure gradually returns to pretreatment levels; rebound hypertension following abrupt withdrawal of the drug has not been reported to date. /Enalaprilat/ For more Absorption, Distribution and Excretion (Complete) data for Enalapril (11 total), please visit the HSDB record page. Metabolism / Metabolites About 60% of the absorbed dose is extensively hydrolyzed to enalaprilat via de-esterification mediated by hepatic esterases. In humans, metabolism beyond bioactivation to enalaprilat is not observed. About 60% of an absorbed dose of enalapril is extensively hydrolyzed to enalaprilat, principally in the liver via esterases. About 20% appears to be hydrolyzed on first pass through the liver; this hydrolysis does not appear to occur in plasma in humans. Enalaprilat is a more potent angiotensin converting enzyme inhibitor than enalapril. There is no evidence of other metabolites of enalapril in humans, rats, or dogs. However, a despropyl metabolite of enalaprilat was identified in urine in rhesus monkeys, accounting for 13% of an oral dose of enalapril maleate. Hydrolysis of enalapril to enalaprilat may be delayed and/or impaired in patients with severe hepatic impairment, but the pharmacodynamic effects of the drug do not appear to be significantly altered. Biological Half-Life The average terminal half life of enalaprilat is 35-38 hours. The effective half life following multiple doses is 11-14 hours. The prolonged terminal half-life is due to the binding of enalaprilat to ACE. Following oral admin, the half-life of unchanged enalapril appears to be <2 hr in healthy individuals and in patients with normal hepatic and renal functions, but may be increased in patients with congestive heart failure. Following oral admin of a single 5 or 10 mg dose of enalapril maleate in patients with congestive heart failure, the half-life of enalapril was 3.4 or 5.8 hr, respectively. Elimination of enalaprilat may also be prolonged in patients with congestive heart failure or impaired hepatic function compared with healthy individuals and patients with hypertension observations of serum concns of enalaprilat over long periods following oral or iv admin suggest that enalaprilat has an avg terminal half-life of about 35-38 hr (range: 30-87 hr). ...The effective half-life for accumulation of enalaprilat (determined from urinary recovery) has been reported to average about 11 hr in healthy individuals with normal renal function. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Enalapril is angiotensin-converting enzyme inhibitor and antihypertensive agent. HUMAN STUDIES: Overdosage of enalapril produces effects that are mainly extensions of the drug's pharmacologic effects as an angiotension converting enzyme inhibitor. Plasma angiotension enzyme activity was completely suppressed within 10-15 hr after acute ingestion of 300-440 mg of enalapril maleate in 2 patients. The most likely manifestation of enalapril overdosage is hypotension, which may be profound and also maybe accompanied by stupor. Onset and duration of the hypotensive effect may be prolonged following acute overdosage. Renal dysfunction, including acute renal failure, hyperkalemia, and hyponatremia may also occur. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue enalapril maleate tablets as soon as possible. ANIMAL STUDIES: There was no evidence of a tumorigenic effect when enalapril was administered for 106 weeks to male and female rats at doses up to 90 mg/kg/day or for 94 weeks to male and female mice at doses up to 90 and 180 mg/kg/day, respectively. There were no adverse effects on reproductive performance of male and female rats treated with up to 90 mg/kg/day of enalapril. Neither enalapril maleate nor the active diacid was mutagenic in the Ames microbial mutagen test with or without metabolic activation. Enalapril was also negative in the following genotoxicity studies: rec-assay, reverse mutation assay with Escherichia coli, sister chromatid exchange with cultured mammalian cells, and the micronucleus test with mice, as well as in an in vivo cytogenic study using mouse bone marrow. Hepatotoxicity Enalapril, like other ACE inhibitors, has been associated with a low rate of serum aminotransferase elevations ( Likelihood score: B (likely but rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because of the low levels of enalapril in breastmilk, amounts ingested by the infant are small and would not be expected to cause any adverse effects in breastfed infants. ◉ Effects in Breastfed Infants None reported in 4 breastfed infants whose mothers were taking oral enalapril 5 to 10 mg daily. ◉ Effects on Lactation and Breastmilk In 15 postmenopausal hypertensive women (prior lactation status not stated), serum prolactin levels were decreased by 22% compared to placebo after enalapril 20 mg once daily for 15 days. The maternal prolactin level in a mother with established lactation may not affect her ability to breastfeed. A woman with pre-eclampsia was treated was started at term with oral enalapril 10 mg daily. Her milk came in on day 3 postpartum and she had no difficulties with nursing during 5 weeks of observation. Protein Binding It is reported that less than 50% of enalaprilat is bound to human plasma proteins, based on limited data from binding studies of enalaprilat in human plasma both by equilibrium dialysis and by ultrafiltration. Interactions When enalapril is administered with diuretics or other hypotensive drugs, the hypotensive effect of enalapril is increased. The effect is usually used to therapeutic advantage, but careful adjustment of dosage is necessary when these drugs are used concomitantly. ... Enalapril and diuretics appear to have additive hypotensive effects; however, severe hypotension and reversible renal insufficiency may occasionally occur, especially in volume and/or sodium depleted patients. Hypotensive drugs that cause release of renin (e.g., diuretics) will increase the hypotensive effect of enalapril. Potassium sparing diuretics (eg, amiloride, spironolactone, triamterene), potassium supplements, or potassium-containing salt substitutes should be used with caution and serum potassium should be determined frequently in patients receiving enalapril, since hyperkalemia may occur. Because ACE inhibitors may promote kinin-mediated prostaglandin synthesis and/or release, concomitant administration of drugs that inhibit prostaglandin synthesis (e.g., aspirin, ibuprofen) may reduce the blood pressure response to ACE inhibitors, including enalapril. Limited data indicate that concomitant administration of ACE inhibitors with nonsteroidal anti-inflammatory agents (NSAIAs) occasionally may result in acute reduction of renal function; however, the possibility cannot be ruled out that one drug alone may cause such an effect. ... Aspirin and other NSAIAs also can attenuate the hemodynamic actions of ACE inhibitors in patients with congestive heart failure. Because ACE inhibitors share and enhance the effects of the compensatory hemodynamic mechanisms of heart failure, with aspirin and other NSAIAs interacting with the compensatory mechanisms rather than with a given ACE inhibitor per se, these desirable mechanisms are particularly susceptible to the interaction and a subsequent potential loss of clinical benefits. As a result, the more severe the heart failure and the more prominent the compensatory mechanisms, the more appreciable the interaction between NSAIAs and ACE inhibitors. Even if optimal dosage of an ACE inhibitor is used in the treatment of congestive heart failure, the potential cardiovascular and survival benefit may not be seen if the patient is receiving an NSAIA concomitantly. In several multicenter studies, concomitant admin of a NSAIA (i.e., a single 350-mg dose of aspirin) in patients with congestive heart failure inhibited favorable hemodynamic effects associated with ACE inhibitors, attenuating the favorable effects of these drugs on survival and cardiovascular morbidity. /ACE inhibitors/ Lithium toxicity has occurred following concomitant administration of enalapril and lithium carbonate and was reversible following discontinuance of both drugs. In one patient, the toxicity was associated with elevated plasma lithium concentration and was manifested as ataxia, dysarthria, tremor, confusion, and altered electroencephalogram, bradycardia and T wave depression also occurred. Moderate renal insufficiency (serum creatinine of 2.2 mg/deciliter) or acute renal failure has also occurred in these patients. The exact mechanism of this interaction remains to be established, but it has been suggested that enalapril may decrease renal elimination of lithium, possibly by increasing sodium excretion secondary to decreased aldosterone secretion or by altering renal function secondary to angiotensin converting enzyme inhibition. Concomitant use of enalapril and some vasodilating agents (eg, nitrates) or anesthetic agents may cause an exaggerated hypotensive response. Patients receiving enalapril concomitantly with nitrates or with anesthetic agents that produce hypotension should be observed for possible additive hypotensive effects. Fluid volume expansion can correct hypotension during surgery or anesthesia if it is thought to result from an enalapril-induced inhibition of the angiotensin II formation that occurs secondary to compensatory renin release. Non-Human Toxicity Values LD50 Mouse oral 2000-3500 mg/kg /Enalapril maleate/ LD50 Rat male oral 2000-3500 mg/kg /Enalapril maleate/ LD50 Rat female oral 2000-3000 mg/kg /Enalapril maleate/ LD50 Rat male sc 1750 mg/kg /Enalapril maleate/ For more Non-Human Toxicity Values (Complete) data for Enalapril (10 total), please visit the HSDB record page. |
| 参考文献 | |
| 其他信息 |
Therapeutic Uses
Angiotensin-Converting Enzyme Inhibitors; Antihypertensive Agents /CLINICAL TRIALS/ ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world. The Web site is maintained by the National Library of Medicine (NLM) and the National Institutes of Health (NIH). Each ClinicalTrials.gov record presents summary information about a study protocol and includes the following: Disease or condition; Intervention (for example, the medical product, behavior, or procedure being studied); Title, description, and design of the study; Requirements for participation (eligibility criteria); Locations where the study is being conducted; Contact information for the study locations; and Links to relevant information on other health Web sites, such as NLM's MedlinePlus for patient health information and PubMed for citations and abstracts for scholarly articles in the field of medicine. Enalapril is included in the database. Enalapril maleate tablets are indicated for the treatment of hypertension. Enalapril maleate tablets are effective alone or in combination with other antihypertensive agents, especially thiazide-type diuretics. The blood pressure lowering effects of enalapril maleate tablets and thiazides are approximately additive. /Included in US product label/ Enalapril maleate tablets are indicated for the treatment of symptomatic congestive heart failure, usually in combination with diuretics and digitalis. /Included in US product label/ For more Therapeutic Uses (Complete) data for Enalapril (9 total), please visit the HSDB record page. Drug Warnings /BOXED WARNING/ When pregnancy is detected, discontinue enalapril maleate tablets as soon as possible. Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus. The most frequent adverse cardiovascular effect of enalapril or enalaprilat is hypotension (including postural hypotension and other orthostatic effects), which occurs in about 1-2% of patients with hypertension and in about 5-7% of those with heart failure, following an initial dose or during extended therapy. Syncope occurred in approximately 0.5 or 2% of patients with hypertension or heart failure, respectively. Hypotension or syncope has required discontinuance of therapy in about 0.1 or 2% of patients with hypertension or heart failure, respectively, receiving enalapril. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue enalapril maleate tablets as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimester of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus. Angioedema may occur, especially following the first dose of enalapril, and, if associated with laryngeal edema, may be fatal. If laryngeal stridor or angioedema of the face, extremities, lips, tongue, or glottis occurs, enalapril should be discontinued and the patient carefully observed until swelling disappears. If swelling is confined to the face and lips, the condition generally responds without treatment; however, antihistamines may provide symptomatic relief. Swelling of the tongue, glottis, or larynx may cause airway obstruction, and appropriate therapy (eg, epinephrine, maintenance of patent airway) should be initiated immediately. Patients should be informed that swelling of the face, eyes, lips, or tongue or difficulty in breathing may be signs and symptoms of angioedema, and that they should discontinue enalapril and notify their physician immediately if any of these conditions occurs. The possibility that patients with a history of angioedema unrelated to angiotensin converting enzyme inhibitors may be at increased risk of developing angioedema while receiving the drugs should be considered. Enalapril is contraindicated in patients with a history of angioedema related to angiotensin converting enzyme inhibitor therapy. Enalapril also is contraindicated in patients with known hypersensitivity to the drug or any ingredient in the formulation. For more Drug Warnings (Complete) data for Enalapril (28 total), please visit the HSDB record page. Pharmacodynamics Enalapril is an antihypertensive agent that exhibits natriuretic and uricosuric properties. Enalapril lowers blood pressure in all grades of essential and renovascular hypertension, and peripheral vascular resistance without causing an increase in heart rate. Individuals with low-renin hypertensive population were still responsive to enalapril. The duration of hypertensive effect in the systolic and diastolic blood pressure persists for at least 24 hours following initial administration of a single oral dose, and repeated daily administration of enalapril confers an additional reduction in blood pressure and a steady-state antihypertensive response may take several weeks. In patients with severe congestive heart failure and inadequate clinical response to conventional antihypertensive therapies, treatment with enalapril resulted in improvements in cardiac performance as observed by a reduction in both preload and afterload, and improved clinical status long-term. Furthermore, enalapril was shown to increase cardiac output and stroke volume while decreasing pulmonary capillary wedge pressure in patients with congestive heart failure refractory to conventional treatment with digitalis and diuretics. In clinical studies, enalapril reduced left ventricular mass, and did not affect cardiac function or myocardial perfusion during exercise. Enalapril is not highly associated with the risk of bradycardia unlike most diuretics and beta-blockers and it does not produce rebound hypertension upon discontinuation of therapy. Enalapril is not reported to produce hypokalaemia, hyperglycaemia, hyperuricaemia or hypercholesterolaemia. In the kidneys, enalapril was shown to increase renal blood flow and decrease renal vascular resistance. It also augmented the glomerular filtration rate in patients with a glomerular filtration rate less than 80 mL/min. When used in combination, enalapril was shown to attenuate the extent of drug-induced hypokalemia caused by hydrochlorothiazide and the antihypertensive effects of both drugs were potentiated. Enalapril Maleate (MK-421) is a prodrug angiotensin-converting enzyme inhibitor (ACEI) that is metabolized in vivo to its active form, enalaprilat, which exerts pharmacological effects by inhibiting ACE. - In the high-methionine-induced endothelial dysfunction model, Enalapril Maleate (MK-421) improved endothelial function not only through inhibiting ACE (reducing angiotensin II synthesis) but also by reducing oxidative stress (downregulating p47phox) and enhancing NO bioavailability (upregulating eNOS expression). - Compared with captopril (another ACEI with a sulfhydryl group), Enalapril Maleate (MK-421) (without a sulfhydryl group) still exhibited significant improvement in endothelial dysfunction, indicating that the sulfhydryl group is not essential for ACE inhibitors to ameliorate endothelial dysfunction [1] |
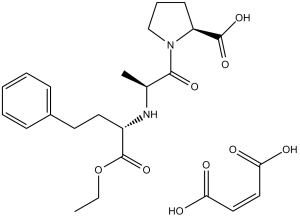
| 分子式 |
C20H28N2O5.C4H4O4
|
|
|---|---|---|
| 分子量 |
492.52
|
|
| 精确质量 |
492.21
|
|
| CAS号 |
76095-16-4
|
|
| 相关CAS号 |
Enalapril-d5 maleate;349554-02-5;Enalapril;75847-73-3
|
|
| PubChem CID |
5388962
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 沸点 |
0ºC
|
|
| 熔点 |
143-144.5ºC
|
|
| 闪点 |
0°C
|
|
| LogP |
1.645
|
|
| tPSA |
170.54
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
10
|
|
| 重原子数目 |
27
|
|
| 分子复杂度/Complexity |
519
|
|
| 定义原子立体中心数目 |
3
|
|
| SMILES |
CCOC(=O)[C@H](CCC1=CC=CC=C1)N[C@@H](C)C(=O)N2CCC[C@H]2C(=O)O
|
|
| InChi Key |
OYFJQPXVCSSHAI-QFPUQLAESA-N
|
|
| InChi Code |
InChI=1S/C20H28N2O5.C4H4O4/c1-3-27-20(26)16(12-11-15-8-5-4-6-9-15)21-14(2)18(23)22-13-7-10-17(22)19(24)25;5-3(6)1-2-4(7)8/h4-6,8-9,14,16-17,21H,3,7,10-13H2,1-2H3,(H,24,25);1-2H,(H,5,6)(H,7,8)/b;2-1-/t14-,16-,17-;/m0./s1
|
|
| 化学名 |
(Z)-but-2-enedioic acid;(2S)-1-[(2S)-2-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]propanoyl]pyrrolidine-2-carboxylic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0304 mL | 10.1519 mL | 20.3037 mL | |
| 5 mM | 0.4061 mL | 2.0304 mL | 4.0607 mL | |
| 10 mM | 0.2030 mL | 1.0152 mL | 2.0304 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
PROACT: Can we Prevent Chemotherapy-related Heart Damage in Patients With Breast Cancer and Lymphoma?
CTID: NCT03265574
Phase: Phase 3 Status: Completed
Date: 2024-02-20
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
