| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |
|

| 靶点 |
IRE1 RNase (IC50 = 0.29 μM)
|
|---|---|
| 体外研究 (In Vitro) |
所有乳腺癌细胞系的生长均被 MKC8866(20 μM;6 天)抑制 [2]。当应用 MKC8866(20 μM;48 小时)时,细胞进入 S 期的频率较低 [2]。在标准情况下,MKC8866(0.2-10 μM;3 天)对所有四种细胞系的活力均表现出剂量依赖性抑制,其中在 LNCaP 细胞中观察到的效果最大 [1]。在 72 小时的持续时间内,MKC8866 (20 μM) 足以完全阻止 NSC 125973 诱导的 XBP1 表达 [1]。
|
| 体内研究 (In Vivo) |
当 NSC 125973 停止时,MKC8866(口服;300 mg/kg;持续 28 天)会减少肿瘤再生 [1]。
MKC8866在多种临床前小鼠模型中单药治疗可显著抑制前列腺癌(PCa)肿瘤生长,并与现有PCa药物具有协同抗肿瘤作用[1] MKC8866在所有测试的PCa细胞系中强烈抑制异种移植肿瘤的生长(图2a)。与对照组相比,MKC8866治疗的肿瘤中XBP1s的表达显著降低,这证实了MKC8866在携带肿瘤的小鼠中具有活性,并且体内IRE1α活性被适当抑制(Supplementary Fig. 4a)。此外,PCNA表达降低,cleaved Caspase-3水平升高,表明MKC8866处理分别导致细胞增殖降低,细胞凋亡增加(补充图4b)。在治疗过程中去除MKC8866导致XBP1s水平反弹,肿瘤生长增强(图2b和补充图4c),表明持续应用MKC8866对其生长抑制作用的重要性。这些结果表明,IRE1α在临床前小鼠PCa模型中具有有效的抗肿瘤作用[1]。 MKC8866与恩杂鲁胺合用抑制肿瘤生长具有较强的协同作用。MKC8866与醋酸阿比特龙和卡巴他赛联合用药也能协同抑制肿瘤生长。综上所述,这些数据表明,在临床前模型中,MKC8866与目前临床上使用的一些中心PCa药物方案具有协同作用。[1] MKC8866增强紫杉醇在体内的有效性[2] 为了确定MKC8866在体内的治疗效果,我们在胸腺裸鼠体内建立了MDA-MB-231肿瘤异种移植物。一旦肿瘤达到可触及的大小(225-250 mm3),将动物随机分为治疗组,分别使用载体、单独300 mg kg - 1 MKC8866、单独10 mg kg - 1紫杉醇或紫杉醇和MKC8866联合治疗。所有组均给予治疗,直至肿瘤达到最大尺寸(2000 mm3)或第60天,以先到者为准。MKC8866在连续口服60次后耐受性良好,并且基于药代动力学异速测量,全身暴露远高于预期的临床治疗水平。与单纯对照相比,单独使用MKC8866治疗没有减弱肿瘤生长(图7a)。对MKC8866治疗的肿瘤中XBP1 mRNA剪接百分比的分析证实了IRE1 RNase活性的降低,验证了靶效应(图7b)。在紫杉醇治疗抑制肿瘤生长的同时,与MKC8866联合治疗可显著增强紫杉醇的疗效。在60天的实验中,与单独使用紫杉醇相比,接受紫杉醇- mkc8866联合治疗的动物的肿瘤生长明显降低(P≤0.0001)(图7c)。与单独使用紫杉醇相比,在第14天(或~700 mm3肿瘤体积)(P≤0.001)或第28天(或~1300 mm3肿瘤体积)(P≤0.05)开始使用紫杉醇- mkc8866联合治疗后观察到类似的协同效应(图7c)。对肿瘤中XBP1剪接的检查显示,紫杉醇治疗增加了IRE1 RNase活性,而与MKC8866联合使用后,IRE1 RNase活性降低(图7d,补充图5)。紫杉醇与MKC8866联合使用后,肿瘤体积的减小也转化为生存期的增加。从第1天到第60天、第14天到第60天、第28天到第60天,每天接受MKC8866联合紫杉醇治疗的小鼠的生存期明显长于单独接受紫杉醇治疗的小鼠(图7e)。 |
| 酶活实验 |
荧光素酶报告基因试验[1]
LNCaP或293T细胞在六孔板中培养,收获前用1 μg pGL3-MYC荧光素酶报告质粒加指定浓度的空载体(pCDNA3)或pCDNA3- flag - xbp1s质粒转染24 h。荧光素酶活性测定采用荧光素酶测定系统和Wallac victor21420 Multilabel计数器。 |
| 细胞实验 |
细胞增殖测定[2]
细胞类型: MCF7、SKBR3、MDA-MB-231 和 MCF10A 细胞 测试浓度: 20 μM 孵育持续时间:6天 实验结果:所有乳腺癌细胞系的增殖均减少。 细胞周期分析[2] 细胞类型: MDA-MB-231、MCF7 和 SKBR3 细胞 测试浓度: 20 μM 孵育持续时间:48小时 实验结果:进入S期的细胞数量减少。 细胞周期分析[1] 细胞类型: LNCaP、VCaP、22Rv1 和 C4-2B 细胞 测试浓度: 0.2, 0.5、1、5、10 μM 孵育时间: 3 天 实验结果: 在一种剂量依赖性方法中抑制所有四种细胞系的活力。 细胞周期分析 [2] 细胞类型: MDA-MB-231 细胞 测试浓度: 20 μM 孵育持续时间:72小时 实验结果:完全阻断 NSC 125973 诱导的 XBP1 表达。 |
| 动物实验 |
Animal/Disease Models: MDA-MB-231 tumor in female athymic nude mice [1]
Doses: 300 mg/kg Route of Administration: oral; continued for 28 days Experimental Results: NSC 125973 diminished tumor regeneration after drug withdrawal. In vivo MDA-MB-231 xenograft model[2] Mice were administered 10 mg kg−1 paclitaxel weekly by intravenous injection. The IRE1 inhibitor, MKC8866, was administered at a dose volume of 10 ml kg−1 from a 30 mg ml−1 suspension in 1% microcrystalline cellulose in a simple sugar at 300 mg kg−1 daily by oral gavage (Vehicle 2). Treatment groups were as follows: For Group 1, the paclitaxel vehicle was administered intravenously weekly and the MKC8866 vehicle was administered orally daily throughout the course of the study. For Groups 2–6, paclitaxel was administered weekly throughout the course of the study. In combination with paclitaxel, MKC8866 was also administered orally daily from day 1 to 28 (Group 3), from day 14 to 60 (Group 4), from day 28 to 60 (Group 5), and from day 1 to 60 (Group 6). Treatments in all groups were administered until tumors reached maximal size or day 60, whichever came first.[2] MKC8866 was administered daily for 28 days at a dose volume of 10 ml kg−1 from a 30 mg ml−1 suspension in 1% microcrystalline cellulose in a simple sugar at 300 mg kg−1 daily by oral gavage (Vehicle 2). Group 1 received paclitaxel (7.5 mg kg−1) alone while Group 2 received paclitaxel (7.5 mg kg−1) plus 300 mg kg−1 MKC8866. After palpable tumors appeared, the mice were randomly grouped and daily received either 300 mg kg−1 MKC8866 or vehicle (1% microcrystalline in 1 g ml−1 sucrose). For the combinatorial experiment of MKC8866 and enzalutamide, mice were assigned into four groups (n = 6 per group): oral gavage of 300 mg kg−1 MKC8866 every other day, oral gavage of 30 mg kg−1 enzalutamide every other day, a combination of daily gavage of either MKC8866 or enzalutamide, and daily gavage of either MKC8866 or 0.5% HP methyl cellulose with 0.1% Tween 20 as a vehicle control. The four groups (n = 6 per group) for the combination of MKC8866 and abiraterone acetate were: oral gavage of 300 mg kg−1 MKC8866 every other day, oral gavage of 20 mg kg−1 abiraterone acetate every other day, a combination of two treatments, and corresponding vehicles. The four groups (n = 6 per group) for the combination of MKC8866 and cabazitaxel were: oral gavage of 300 mg kg−1 MKC8866 every other day, intraperitoneal injections of 5 mg kg−1 cabazitaxel twice a week, a combination of two treatments, and corresponding vehicles. Tumor weight was measured in the end of experiment.[1] |
| 参考文献 | |
| 其他信息 |
IRE1 RNase Inhibitor ORIN1001 is an orally bioavailable inhibitor of the serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1 (inositol-requiring enzyme 1 alpha; IRE1), with potential immunoactivating, chemosensitizing and antineoplastic activities. Upon oral administration, IRE1 RNase inhibitor ORIN1001 targets and binds to the RNase domain of IRE1, thereby inhibiting the activity of IRE1. This prevents activation of the IRE1/X-Box Binding Protein 1 (XBP1) pathway, inhibits unfolded protein response (UPR) stress adaptation and prevents the production of pro-tumorigenic factors. This may inhibit tumor growth in which IRE1 is overactivated. In addition, ORIN1001 abrogates the immunosuppressive tumor microenvironment (TME) through cytotoxic T-cell infiltration and depletion of immunosuppressive myeloid-derived suppressor cells (MDSCs) in the TME. IRE1, an endoplasmic reticulum (ER)-localized transmembrane protein containing an ER luminal stress-sensing domain and a cytoplasmic facing RNase domain, acts as a key sensor for the UPR and plays a key role in the response to and resolution of ER stress. IRE1 is involved in both protein phosphorylation and mRNA processing and degradation in response to ER stress-dependent signaling. IRE1 is frequently co-amplified with the MYC oncogene.
|
| 分子式 |
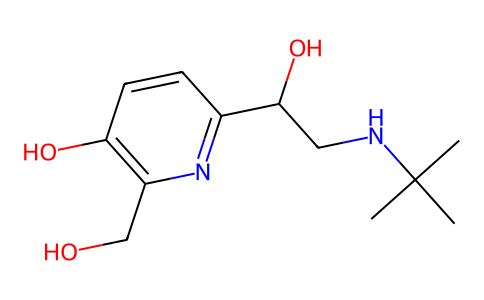
C18H19NO7
|
|---|---|
| 分子量 |
361.3460
|
| 精确质量 |
361.12
|
| 元素分析 |
C, 59.83; H, 5.30; N, 3.88; O, 30.99
|
| CAS号 |
1338934-59-0
|
| PubChem CID |
89542346
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| LogP |
0.5
|
| tPSA |
102Ų
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
611
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
IFDGMRMUJYGWQQ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C18H19NO7/c1-10-11-7-14(24-2)16(22)13(9-20)17(11)26-18(23)12(10)8-15(21)19-3-5-25-6-4-19/h7,9,22H,3-6,8H2,1-2H3
|
| 化学名 |
7-Hydroxy-6-methoxy-4-methyl-3-(2-morpholino-2-oxoethyl)-2-oxo-2H-chromene-8-carbaldehyde
|
| 别名 |
ORIN1001; ORIN-1001; ORIN 1001;
MKC-8866; MKC 8866; 2H-1-Benzopyran-8-carboxaldehyde, 7-hydroxy-6-methoxy-4-methyl-3-(2-(4-morpholinyl)-2-oxoethyl)-2-oxo-; UNII-1NZ0YBP9HB;MKC8866; IRE1-IN-8866; IRE1IN8866; IRE1;
IN 8866; IRE1-IN8866; IRE1-IN 8866; IRE1IN-8866; IRE1IN 8866
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~6.67 mg/mL (~18.46 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7674 mL | 13.8370 mL | 27.6740 mL | |
| 5 mM | 0.5535 mL | 2.7674 mL | 5.5348 mL | |
| 10 mM | 0.2767 mL | 1.3837 mL | 2.7674 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA